This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Livornese — I saw the sign…and the answer is no—FDA-approved labeling apparently is not enough under state failure-to-warn laws, according to certain courts. That requirement would only be fulfilled if FDA agreed to such a change. But we digress. And indeed, after Pliva v.
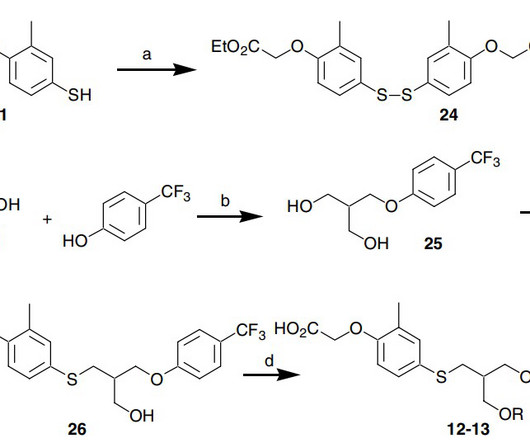
4] Seladelpar was approved for medical use in the United States in August 2024. [1] 2007 Jul 15;17(14):3855-9. Epub 2007 May 10. Seladelpar cas 851528-79-5 C 21 H 23 F 3 O 5 S, 444.47 1] It is used as the lysine dihydrate salt. [1] 1] It is a PPARδ receptor agonist. [1] Seladelpar works to block bile acid synthesis. 2007.05.007.
Food and Drug Administration (FDA) has approved PREVNAR 20 (Pneumococcal 20-valent Conjugate Vaccine) for the prevention of invasive disease and pneumonia caused by the 20 Streptococcus pneumoniae (pneumococcus) serotypes in the vaccine in adults ages 18 years and older. Following today’s FDAapproval, the U.S.
Related news Scientific coalition developing surveillance system for detecting emerging pandemics in real-time While combing through the human genome in 2007, computational geneticist Pardis Sabeti made a discovery that would transform her research career. Finding participants for the study would be challenging too.
FDA advisory committees recommended just 50 percent of the 18 new therapies and indications they reviewed in 2020, the lowest rate since 2007, and the agency seems to be reserving the panels for more problematic applications, according to Prevision Policy, a Washington, D.C.-based based research firm.
” The study looked at more than 301,000 antipsychotic prescriptions filled between 2007 and 2017 for children ages 2 to 7 who were covered by private insurance. . “However, fewer than half of the children receiving antipsychotic treatment in our study had a visit with a psychiatrist or a psychotherapy claim.”
If an FDA-approved carve-out could support an intent to induce infringement claim, the use of the “section viii pathway would be substantially deterred.” Plainly, the Government brief states “The decision below is incorrect.
If successful, this work could result in the repurposing of several FDA-approved therapeutics for the purpose of extending the human lifespan, at a lower cost and over faster timelines than conceivably possible with de novo drug discovery. The Scheibye-Knudsen lab has analyzed 1.5 billion prescriptions from 4.8
Since 2007, they’d been studying Lassa fever, an often deadly illness caused by infection with the Lassa virus, which is endemic in West Africa. Sentinel is also developing a SHINE test to detect Lassa virus in rural clinics in Nigeria, where there are currently no FDA-approved diagnostics for the disease.
Plaintiffs performed the necessary studies on BRAVECTO and filed an NADA on April 8, 2014; FDAapproved the NADA on May 15, 2014. FDA, since 1991, had publicly signaled that the opening of an INAD file starts the testing phase for a new animal drug. FDA also consistently interpreted it that way between 1990 and 2007.
Until recently, the FDA relied on a monograph process through which firms could bring OTC drugs to market without FDAapproval so long as it adhered to pre-set terms under the monograph. The FDA maintains a webpage with the full rulemaking history for OTC nasal decongestant drug products.
1067, the “ Ensuring Timely Access to Generics Act of 2023 ,” and it would fundamentally transform the playing field for NDA, ANDA, BLA, and aBLA applicants seeking to preserve their rights in the wake of an adverse FDAapproval decision. 110-85 (2007), as amended by Section 301 of Pub. That bill is S.
The FDA first proposed a rule on “Prescription Drug Labeling: Medication Guide Requirements” in 1995 , which was finalized in 1998. However, over the last decade the FDA and other stakeholders have acknowledged that there are opportunities for improvement. After all, companies already have FDA-approved labels.
These products are also required to comply with certain labeling expectations and manufacturers are expected to follow a specific set of current good manufacturing practices ( a final rule issued in 2007). Dietary supplement products do not need approval prior to being marketed – unless they contain a new dietary ingredient (NDI).
Plus, many of these kits claimed to be EU or FDAapproved, when they were anything but… Quite simply, they were ILLEGAL, but I was still able to buy them online – and so can you. s one from The Daily Mail in the UK, which was highlighting the risks all the way back in 2007… And very little has changed since then!
As AgencyIQ has previously discussed , the Tropical Disease PRV program was actually the first of FDA’s voucher programs, introduced in 2007 to incentivize the development of products to treat diseases (i.e., In January 2024, the FDAapproved the first-ever importation plan , from the state of Florida.
Effective as of 2007, Medicare has offered coverage for routine costs in clinical trials. FDA-approved IDE devices may be deemed qualifying under the Medicare Benefit Policy Manual, Chapter 14. in 2007, deeming a clinical trial to be qualifying by solely pointing to the seven desirable characteristics below is not recommended.
Zidovudine showed promise against multiple HIV strains in cultured cells, and the Food and Drug Administration (FDA) approved it for human studies within five months. By 1987, the FDA licensed zidovudine after trials showed it increased survival rates.
This “keep selling” theory found some traction in two 2007 decisions, Abigail Alliance for Better Access to Developmental Drugs v. 2007) (en banc), and CareToLive v. Ohio 2007), but has generally failed to expand that foothold since. von Eschenbach , 495 F.3d 3d 695 (D.C. von Eschenbach , 525 F. 2d 952 (S.D.
We’ve been diligently preparing bottom ten annual lists since 2007, even though it’s distasteful, because if we don’t do it nobody else is likely to, and these abominable decisions deserve to be called out for what they are. Holley also allowed a “pre-approval” warning claim to escape preemption, largely on the same rationale.
A hundred years later, we detailed three rounds of litigation over Massachusetts’ serial efforts to ban, or at least substantially limit, the use of FDA-approved pain medications. For instance, Sorsaia primarily involves a challenge to a state trying to prevent in-state use of an FDA-approved drug for its FDA-approved use.
The core premise of Bexis’ article is very simple: Once the FDA has said “yes” and approved a particular drug for a particular indication (“intended use”) for sale in the United States, federal preemption precludes any state from saying say “no” and trying to ban that same FDA-approved drug. In GenBioPro, Inc.
But in prescription medical product liability litigation, products must receive FDAapproval, clearance or other authorization (hereafter, collectively referred to as “approval” for short) before they can be marketed. to determine whether a proposed alternative drug would have received FDAapproval.” at 237-38.
2007 WL 4042757, at *3 (N.D. 15, 2007); McNeil v. Texas has a rebuttable presumption that FDA-approved prescription drug labels are adequate, and Plaintiff here is unable to rebut that presumption. Novartis Pharmaceuticals Corp. , 2d 898, 909-10 (W.D. 2013); Massa v. Genentech Inc. 2012 WL 956192, at *5 (S.D.
2012), addressed a challenge to the application of Idaho’s Pain-Capable Unborn Child Protection Act to criminalize the use of an FDA-approved abortifacient medication obtained through an internet prescription and mailed to the plaintiff from out of state. 124 (2007), which would also be knocked out by Dobbs if it sticks.
After attending a patient education class, seeking a second opinion, being warned that she was at increased risk for revision as a woman, and being warned of a risk of metal ions accumulating in her blood, she went forward with the BHR implant in her left hip on June 25, 2007. at *7 (citing the case discussed here ).
We discussed these concerns – which have since crystallized into what is called either the “municipal cost recovery rule” or the “free public services doctrine” – more detail back in one of the Blog’s early(2007) posts. In other words, the generic manufacturers are not allowed to change the FDA-approved label.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.































Let's personalize your content