This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
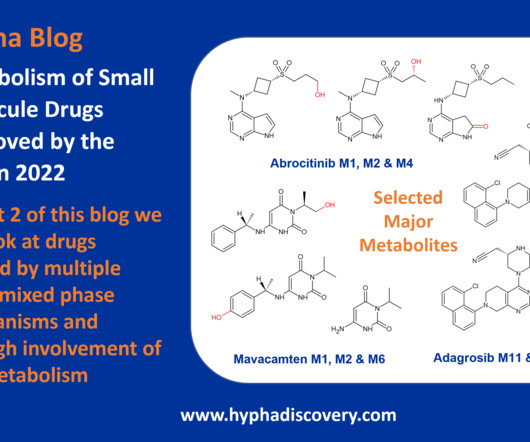
Metabolism of 2022 FDAapproved small molecule drugs part 2 Mixing it Up By Julia Shanu-Wilson In Part 1 of this topic we looked at metabolism of the small molecule drugs approved by the FDA in 2022 that were mediated by CYP3A4. 2008 Mar;153 Suppl 1(Suppl 1):S82-9. Dermavant’s tapinarof is one such friend.
First and only FDA-approved subcutaneous treatment option for anti-aquaporin-4 antibody positive NMOSD that can be self-administered by a person with NMOSD or a caregiver every four weeks. First and only approved therapy for NMOSD designed to target and inhibit interleukin-6 receptor activity, using novel recycling antibody technology.
Livornese — I saw the sign…and the answer is no—FDA-approved labeling apparently is not enough under state failure-to-warn laws, according to certain courts. The GAO Report further explained that the agency did not have the resources to regulate the estimated 100,000 OTC drugs marketed through the monograph process.
This major shift to the orthodox tradition of using animal experiments in drug testing dates back the Aristotle’s time and cemented 80 years ago with initial federal mandate of drug safety regulation of 1938. To this end, the FDA’s newly created iSTAND initiative drives the path toward regulatory approval for devices like organ-on-chips.
156, a patent may be extended only once (even if it would be eligible for extension on more than one occasion because it applies to several FDA-approved products), and only one patent may be extended for each regulatory review period. Part 1: Multiple PTEs Under the PTE statute at 35 U.S.C. § To that end, 35 U.S.C. § 156(c)(4)
John Hospital, researchers compared 649 patients from two cohorts: a recent cohort (2017–2019) from the RECOVER III post-market approval (PMA) study, after the widespread adoption of the best practice of placing Impella pre-PCI, and a cohort from before PMA (2008–2014) when the practice of placing Impella pre-PCI was not yet widely adopted.
The FDA recently concluded its work on a proposed rule focused on PMI. The regulator sent the rule to the White House’s Office of Information and Regulatory Affairs (OIRA) on October 4, 2022. The content of the PMI : The regulation describes, in broad terms, what must be included in the PMI.
1067, the “ Ensuring Timely Access to Generics Act of 2023 ,” and it would fundamentally transform the playing field for NDA, ANDA, BLA, and aBLA applicants seeking to preserve their rights in the wake of an adverse FDAapproval decision. 110-316 (2008) and by Section 1135 of the FDA Safety and Innovation Act (“FDASIA”), Pub. —The
temporary exceptions from some of the requirements of the Ryan Haight Act of 2008) are extended for an additional year, until December 31, 2025. Thus, while continuing to review industry feedback, DEA is still working on promulgating a more workable final set of telemedicine regulations. The federal telemedicine flexibilities (i.e.,
FDA , 78 F.4th 2023), was the Fifth Circuit’s blatantly politicized attack on the FDA’sregulation of abortion-related drugs. That’s significant because the labels for over 500 drugs already have such information, under a voluntary FDA program. Remember Riegel ( 2008+1 )? 4th 210 (5th Cir. Gilead Sciences, Inc.
Constitution’s Supremacy Clause, it strikes a balance between state and federal power on issues of public health and safety, and raises questions about whether our country’s approach to regulation and litigation makes sense for pharmaceuticals and medical devices (spoiler alert: it doesn’t). 312 (2008), and considering 21 U.S.C.
The ASR program was initially open to regulated manufacturers of twelve device categories; Intravascular (I.V.) FDA, CDER, “Guidance for Industry − Medical Device Reporting − Alternative Summary Reporting (ASR) Program, 2000 WL 34503093, at *1 (Oct. 312 (2008). an exemption from the individual event reporting requirements.”
As evidenced by our PMA Preemption Score Card , on which today’s case became the 651 st entry, defendant manufacturers of FDA-approved Class III medical devices generally do pretty well with preemption motions. 312 (2008). Since that would encroach on the FDA’s regulatory authority, this claim is also expressly preempted.
2015), finally gave appellate recognition to the preemption of design defect claims for FDA-approved branded prescription drugs. FDAapproved the drug with its particular formulation and the manufacturer could not have changed the formulation on its own. As detailed here , the decision in Yates v. 3d 281 (6th Cir.
But in prescription medical product liability litigation, products must receive FDAapproval, clearance or other authorization (hereafter, collectively referred to as “approval” for short) before they can be marketed. Lederle Laboratories , 2008 WL 972657 (W. In Bruesewitz v. Wyeth LLC , 562 U.S. at 237-38. 2d at 401.
440 (2008) Buckman was not cited at all in the Merck Sharp & Dohme Corp. Plaintiffs Legal Committee , 531 U.S. Kent , 552 U.S. Albrecht , 139 S. 2019), or Mutual Pharmaceutical Co. Bartlett , 570 U.S. 472 (2013), implied preemption decisions, cited only by the dissent in Wyeth v. . Jones , 225 U.S. Davidowitz , 312 U.S. Myrick , 514 U.S.
When you hear Class III medical device product liability case, you should look for all claims to be dismissed unless there is something as unusual as a basis to claiming the plaintiff’s particular device deviated from its FDA-approved specifications. An unfortunate fiction developed post- Buckman —particularly after Riegel v.
June 24, 2022), we fully expect attempts by such states to ban FDA-approved prescription drugs that can be used to bring about abortions by chemical means. On occasion, states have attempted to prohibit sale and/or use of non-abortion-related FDA-approved products, but with one exception, these efforts have not resulted in litigation.
2012), addressed a challenge to the application of Idaho’s Pain-Capable Unborn Child Protection Act to criminalize the use of an FDA-approved abortifacient medication obtained through an internet prescription and mailed to the plaintiff from out of state. See this post on one of our decisions from 2008.) Hiedeman, 694 F.3d
Even before the anti-osteoporosis drug Fosamax was FDAapproved, its manufacturer was aware of a biologically plausible mechanism for that class of drug (“bisphosphonates”) to cause low-energy – later renamed “atypical” ? The manufacturer informed the FDA, but no evidence then established that risk as anything more than hypothetical.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.

























Let's personalize your content