This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
As with other FDA-regulated products, such as human drugs and medical devices, the “regulatory review period” is composed of a “testing phase” and a “review phase.” The “review phase” is the period between the initial submission and approval of the NADA. FDA’s PTE regulations at 21 C.F.R. When the U.S.
WAKIX is the first and only treatment approved by the FDA for people with excessive daytime sleepiness or cataplexy associated with narcolepsy that is not scheduled as a controlled substance by the U.S. WAKIX received FDAapproval for the treatment of excessive daytime sleepiness in adult patients with narcolepsy in August 2019.
This major shift to the orthodox tradition of using animal experiments in drug testing dates back the Aristotle’s time and cemented 80 years ago with initial federal mandate of drug safety regulation of 1938. The first successful chip adaptation to a lung model was first described in 2010 by Donald Ingber, a bioengineer at Wyss institute.
Karst — While the Biologics Price Competition and Innovation Act (“BPCIA”) is inherently distinct from the Hatch-Waxman Act, many of the fundamental concepts FDA adopted as it enacted the Hatch-Waxman Act made their way into FDA’s implementation of the BPCIA. FDA explained that its bioequivalence regulations at 21 C.F.R.
156, a patent may be extended only once (even if it would be eligible for extension on more than one occasion because it applies to several FDA-approved products), and only one patent may be extended for each regulatory review period. Another case of same-day (and same-time) FDAapprovals! To that end, 35 U.S.C. § 156(c)(4)
The FDA recently concluded its work on a proposed rule focused on PMI. The regulator sent the rule to the White House’s Office of Information and Regulatory Affairs (OIRA) on October 4, 2022. The content of the PMI : The regulation describes, in broad terms, what must be included in the PMI.
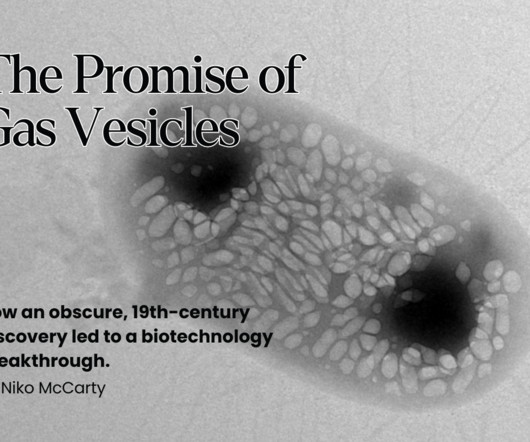
For more than a century, scientists thought of gas vesicles as little more than a natural curiosity and, later, as a way for microbes to regulate buoyancy. Right) The passage of text from a 2010 review that inspired Mikhail Shapiro to engineer gas vesicles. Voigt, Biotechnology Journal (2010). 31,500x magnification.
BY CHELSEY MCINTYRE, PHARMD | JUN 3, 2024 8:43 PM CDT Regulatory background: DSHEA and dietary supplements The Dietary Supplement Health and Education Act ( DSHEA ) of 1994 defines the FDA’s authority in the regulation of dietary supplement products and dietary ingredients. See AgencyIQ’s analysis of the draft guidance here. ]
This was the founding ethos of Moderna, the mRNA vaccine developer, when it was launched in 2010 by Noubar Afeyan and Doug Cole at Flagship Pioneering. In response to this hysteria, regulation was aggressively tightened. An alternative approach is to make molecules not in tubes, vials, or cells, but inside the body itself.
Food and Drug Administration (FDA) approved LUMAKRAS for the treatment of adult patients with KRAS G12C-mutated locally advanced or metastatic NSCLC, as determined by an FDA-approved test, who have received at least one prior systemic therapy. In May, the U.S. Bemarituzumab. Tarlatamab (AMG 757). AMG 451 / KHK4083.
BY RACHEL COE, MSC | APR 24, 2024 10:56 PM CDT Biosimilars and interchangeable products The FDA was first authorized to develop a pathway for the approval of biosimilar products in 2010 under the Biologics Price Competition and Innovation (BPCI) Act. Yes, the guidance confirms.
IMBRUVICA is the only FDA-approved medicine in WM and cGVHD. IMBRUVICA was one of the first medicines to receive FDAapproval via the Breakthrough Therapy Designation pathway. 2010; 116(20): 4777–4787. BTK signaling is needed by specific cancer cells to multiply and spread. Accessed May 2021. 3 Shanafelt, et al.
Biosimilars are biological products that are highly similar (but not identical) to a previously approved biological product and have “no clinically meaningful” differences relative to the original reference product. Developing biosimilars is an extensive and expensive process.
IMBRUVICA is the only FDA-approved medicine in WM and cGVHD. IMBRUVICA was one of the first medicines to receive FDAapproval via the Breakthrough Therapy Designation pathway. 2010; 116(20): 4777–4787. BTK signaling is needed by specific cancer cells to multiply and spread. Accessed May 2021. 4 Shanafelt, et al.
As the DDL blog has previously reported , Michigan’s longstanding presumption of non-defectiveness applicable to FDA-approved drugs was recently repealed by the Michigan legislature in S.B. As this provision is newly applicable to FDA-approved products, it has not been tested by the courts on this issue. See Prasol v.
FDA , 78 F.4th 2023), was the Fifth Circuit’s blatantly politicized attack on the FDA’sregulation of abortion-related drugs. 23, 2010) ( here ), can supersede our list, so far they’ve been thankfully uncommon. Holley also allowed a “pre-approval” warning claim to escape preemption, largely on the same rationale.
But in prescription medical product liability litigation, products must receive FDAapproval, clearance or other authorization (hereafter, collectively referred to as “approval” for short) before they can be marketed. to determine whether a proposed alternative drug would have received FDAapproval.” at 237-38.
When you hear Class III medical device product liability case, you should look for all claims to be dismissed unless there is something as unusual as a basis to claiming the plaintiff’s particular device deviated from its FDA-approved specifications. An unfortunate fiction developed post- Buckman —particularly after Riegel v.
Even before the anti-osteoporosis drug Fosamax was FDAapproved, its manufacturer was aware of a biologically plausible mechanism for that class of drug (“bisphosphonates”) to cause low-energy – later renamed “atypical” ? At the FDA’s suggestion, the manufacturer withdrew that supplement, but kept trying. femur fractures.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.























Let's personalize your content