This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Rescheduling out of schedule I would allow for the medical use of FDA-approved prescription drugs dispensed by DEA-registered, state licensed pharmacies pursuant to prescriptions issued by similarly DEA-registered, state licensed practitioners. Department of Justice (“DOJ”) and DEA respond?
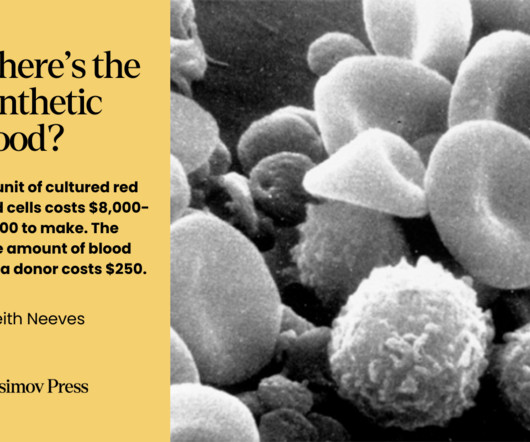
Biotechnology Journal (2013). Despite the initial promise of PFCs and FDAapproval of a product called Fluosol-DA in 1989, PFCs have many drawbacks. A cost analysis to make one unit of red blood cells from stem cells. Data from Rousseau G.F. Each particle includes a lipid shell and a liquid core containing biomolecules.
Palmer — Last week FDA published a long-awaited Draft Guidance for outsourcing facilities addressing the Prohibition on Wholesaling Under Section 503B of the Federal Food, Drug, and Cosmetic Act (Draft Guidance). See 21 U.S.C. 353b(a). Section II at 2. Draft Guidance III.B.2(e)
FDA addressed the expanding practice of drug compounding in 1992 by issuing a compliance policy guide that clarified that pharmacies which compounded products at certain scales, for certain purposes, or without FDAapproval were clearly operating “outside the bounds of traditional pharmacy practice.”
The sponsor-initiated IND is conducted by a pharma company seeking commercial approval. Q: When you want to do a study of an FDA-approved drug under an investigational indication, is an IND still required or can you seek an exemption? Then you can submit an application for approval. of this guidance. of this guidance.
Curiously, there is no direction for the patient to seek out the full FDA-approved labeling. Information included in the PMI : FDA intends that the PMI would “highlight the most important information that patients need to know to help them use their prescription drug products safely and effectively.”
Such a situation is commonplace in the clinical trial realm, in which investigational drug products which are not already FDAapproved are administered to patients. Through this program, the FDA can conduct periodic stability testing on select products and extend the labeled shelf life accordingly. Enter the beyond-use date.
In addition, Amarin recognized licensing and royalty revenue of approximately $1.3 This compares with licensing and royalty revenue of $0.2
Licensing and royalty revenue.
The increase was also driven by VASCEPA sales outside of the United States of approximately $0.5 million and $8.9 million and $5.7 million and $1.1
If FDAapproved, it will join other previously-approved “-gepant” drugs [ rimegepant ] and [ ubrogepant ] as an additional treatment alternative for patients with migraine, particularly those for whom traditional triptan therapy has proven ineffective. and > 300 mg/mL at lower pH pKa 4.8 2021 Jan 1;31:127624.
Zidovudine showed promise against multiple HIV strains in cultured cells, and the Food and Drug Administration (FDA) approved it for human studies within five months. By 1987, the FDAlicensed zidovudine after trials showed it increased survival rates. This has happened before with other drugs.
The FDA requires real science for warnings; thus it had not mandated any warning remotely resembling Prop 65. The plaintiff failed to identify any method by which a generic (or any other) drug manufacturer could add a Prop 65 warning without deviating from FDA-approved labeling, thereby violating federal law. 13) McGee v.
2013); Massa v. Texas, unlike most states, enforces a strong statutory presumption that prescription medical product warnings complying with FDA requirements imposed by “pre-market approval or licensing of the product” are adequate as a matter of law. Novartis Pharmaceuticals Corp. , 2d 898, 909-10 (W.D. Genentech Inc.
2012), addressed a challenge to the application of Idaho’s Pain-Capable Unborn Child Protection Act to criminalize the use of an FDA-approved abortifacient medication obtained through an internet prescription and mailed to the plaintiff from out of state. As we discussed here , McCormack v. Hiedeman, 694 F.3d 3d 1004 (9th Cir.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.



















Let's personalize your content