This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Food and Drug Administration (FDA) to market Ephedrine Sulfate Injection in a ready-to-use 50mg/10 ml single use vial presentation. Under an exclusive licensing agreement with Endo International’s (NASDAQ: ENDP) subsidiary, Endo Ventures Limited, Par Pharmaceuticals’ Sterile Products division will launch and distribute the product.
To further facilitate generic drug development, and to assist the generic pharmaceutical industry in this process, the FDA publishes product-specific guidances (PSGs) describing the agency’s current thinking and expectations on how to develop generic drug products that are therapeutically equivalent to their brand name counterparts.
Food and Drug Administration (FDA) approval of the VENTANA MMR RxDx Panel for advanced or recurrent endometrial cancer patients. FDAapproval of the VENTANA MMR RxDx Panel provides clinicians with access to a fully automated, easy-to-use MMR test to identify patients who are eligible for therapy with JEMPERLI.
Founded in 2015, Standigm has raised $23M from leading investors and developed an elite team with multi-disciplinary expertise in chemistry, biology, pharmacology, artificial intelligence, and data structures to ease the pains of patients all over the world.
“By committing to bring biosimilar formulations such as Hyrimoz citrate-free HCF to patients, we are serving a critical need in expanding access to important medicines and fueling pharmaceutical innovation.”. The adalimumab reference medicine (Humira ®* ) was first approved with an adalimumab concentration of 50 mg/mL.
4 For the treatment of rare genetic disorders especially, drugs with genetically supported targets are more than twice as likely to be approved 5 , thereby indicating genetics and genomics can empower companies to develop better drugs. Human genetics evidence supports two-thirds of the 2021 FDA-approved drugs. Epub 2015 Nov 12.
Announcing the first biopharma IP-NFT Transaction Funding the first longevity research project by utilizing intellectual property NFTs in the pharmaceutical space Summary The vote has passed, the decision is made: The Scheibye-Knudsen Lab will be the first research organisation to fund their longevity research via an IPNFT.
According to preliminary data from one Phase 1 and two Phase 2 clinical trials reported by Vertex Pharmaceuticals Inc., The first major advance in molecularly targeted drug treatment for the disease came in 2012, when the Food and Drug Administration (FDA) approved ivacaftor (Kalydeco). About 30,000 Americans have CF.
In addition to the early work of Matt Disney at the Scripps Research Institute and others in the academic community showing that this was even possible, a number of pharmaceutical companies have made important advances towards drugging RNA structures to discover new therapeutics.
The adalimumab reference medicine (Humira ® *) was first approved with an adalimumab concentration of 50 mg/mL. 1 In 2015, the EMA and US FDAapproved Humira ® HCF, which contains adalimumab at a concentration of 100 mg/mL.
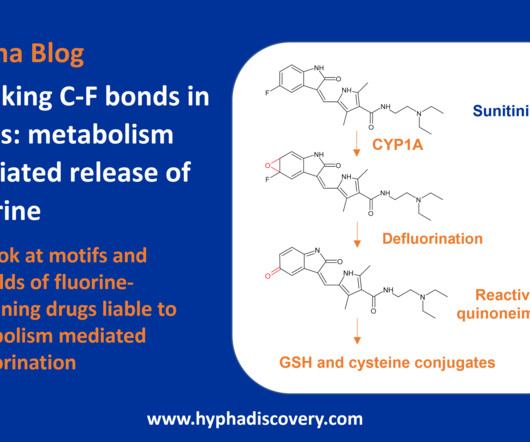
Now >20% of all commercialised medicines in the pharmaceutical industry contain a fluorine atom [2]. In 2021, almost one third of FDAapproved drugs contained at least one fluorine. References [1] Metabolic and Pharmaceutical Aspects of Fluorinated Compounds. Benjamin M. Airaksinen. Blackaby, Alastair J.
The sponsor is the pharmaceutical company conducting the trial. A: Working in a pharmaceutical company is the best way to learn this. The sponsor-initiated IND is conducted by a pharma company seeking commercial approval. A: For FDA 1572 is a Statement of Investigator under US Regulations/Laws. of this guidance.
Food and Drug Administration (FDA) has issued Emergency Use Authorization (EUA) for its single-dose COVID-19 vaccine, developed by the Janssen Pharmaceutical Companies of Johnson & Johnson, to prevent COVID-19 in individuals 18 years of age and older. There is no FDA-approved vaccine to prevent COVID-19.
The recruitment period for the study spanned from December 2015 to July 2017. Food and Drug Administration (FDA) approval for nmCRPC on February 14, 2018 and was approved for mCSPC on September 17, 2019. About the Janssen Pharmaceutical Companies of Johnson & Johnson. ERLEADA ® received U.S.
Similarly, only one product in 2023 was recommended for approval under “ exceptional circumstances ” – Loargys. This approval type has always been uncommon – before the pandemic, this pathway was used just eight times between 2015 and 2019. [ See AgencyIQ’s analysis of this approval type from 2015-2021 here.]
January 29, 2021 – Johnson & Johnson (NYSE: JNJ) (the Company) today announced topline efficacy and safety data from the Phase 3 ENSEMBLE clinical trial, demonstrating that the investigational single-dose COVID-19 vaccine in development at its Janssen Pharmaceutical Companies met all primary and key secondary endpoints. Source link.
FDA addressed the expanding practice of drug compounding in 1992 by issuing a compliance policy guide that clarified that pharmacies which compounded products at certain scales, for certain purposes, or without FDAapproval were clearly operating “outside the bounds of traditional pharmacy practice.”
Such a situation is commonplace in the clinical trial realm, in which investigational drug products which are not already FDAapproved are administered to patients. Under the Pandemic and All-Hazards Preparedness Reauthorization Act of 2013 (PAHPRA) , the FDA also has some authority to extend MCM expiration dates.
While stimulant use disorder is increasing, there are currently no FDA-approved medications. According to the Substance Abuse and Mental Health Services Administration (SAMHSA), “between 2008 and 2015, amphetamine-related hospitalizations more than tripled, increasing from 55,447 instances to 206,180.
In July 2014, Opdivo was the first PD-1 immune checkpoint inhibitor to receive regulatory approval anywhere in the world. Opdivo is currently approved in more than 65 countries, including the United States, the European Union, Japan and China. 1%) as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations.
An update on the Medical Device Quality Management System Regulation : In February 2022, the FDA proposed a new rule which would effectively transition the agency away from its longstanding Quality System Regulation (QSR) in favor of the ISO 13485:2016 standard. Read our analysis of that rule here and here. ]
To implement that authority, FDA published a final rule in the Federal Register on September 15, 2015 [80 FR 55237] which revised 21 CFR 1.94 to provide notice and an opportunity for owners or consignees of the drug to appear before the Agency and introduce testimony prior to the destruction of their drug.
Date What’s Happening Explanation Source October 31 FDA deadline on Florida drug importation plan The FDA has said in court filings that it plans to make a ruling on Florida’s prescription drug importation plan by October 31. The following PDUFA dates were obtained from publicly available sources.
In July 2014, Opdivo was the first PD-1 immune checkpoint inhibitor to receive regulatory approval anywhere in the world. Opdivo is currently approved in more than 65 countries, including the United States, the European Union, Japan and China. 1%) as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations.
If FDAapproved, it will join other previously-approved “-gepant” drugs [ rimegepant ] and [ ubrogepant ] as an additional treatment alternative for patients with migraine, particularly those for whom traditional triptan therapy has proven ineffective. Migraine is both widespread and disabling. New Haven, CT.
12/29/2023 FDORA, Section 3210 Accelerated Approval : FDA’s Accelerated Approval Council must publish a report within one year of the passage of the FDORA legislation regarding its annual activities. ” The report will also cover Federal agency roles in addressing vulnerabilities and statutory limitations.
14, 2020 /PRNewswire/ — Regeneron Pharmaceuticals, Inc. Food and Drug Administration (FDA) approved Inmazeb (atoltivimab, maftivimab and odesivimab-ebgn) for the treatment of infection caused by Zaire ebolavirus in adult and pediatric patients, including newborns of mothers who have tested positive for the infection.
Society and culture Legal status Treosulfan was authorized for medical use in the European Union in June 2019, [9] and approved for medical use in the United States in January 2025. [11] Retrieved 2 January 2023. ^ “Trecondi (Link Medical Products Pty Ltd T/A Link Pharmaceuticals)” Therapeutic Goods Administration (TGA).
Although Regeneron Pharmaceuticals has been in the news lately for its antibody cocktail against COVID-19 that was used to treat President Trump, it’s not the only arrow in the company’s quiver. Food and Drug Administration (FDA) approved Inmazeb, a three-antibody cocktail to treat Ebola infections in adults and children.
Instead of highlighting the cognitive dissonance of the possibility that both of these theories of liability could be viable, we could be touting the creativity of the plaintiff lawyers trying different approaches to force pharmaceutical companies to play an unfun version of Simon Says.
As the DDL blog has previously reported , Michigan’s longstanding presumption of non-defectiveness applicable to FDA-approved drugs was recently repealed by the Michigan legislature in S.B. As this provision is newly applicable to FDA-approved products, it has not been tested by the courts on this issue.
Another of our posts quoted similar concerns raised by our clients in the pharmaceutical industry as the matter was being successfully appealed to the United States Supreme Court: The Fifth Circuit’s ruling threatens to stifle pharmaceutical innovation by disrupting industry’s reasonable investment-backed expectations. Longbons T.,
2023) (HHS cannot force pharmaceutical manufacturers to sell unlimited amounts of prescription drugs at a discount) ( here ). The FDA requires real science for warnings; thus it had not mandated any warning remotely resembling Prop 65. Forest Pharmaceuticals, Inc. Pacira Pharmaceuticals, Inc. 4th 851 (6th Cir.
Ortho-McNeil-Janssen Pharmaceuticals, Inc. , 2015), and the Supreme Court’s opinion in Mutual Pharmaceutical Co. It makes no difference in our view that the FDA ultimately approved TAF-based drugs. It is just a claim that the defendant should have proposed to the FDA a different warning than what the FDAapproved.
in part to aid the cause of medical device and pharmaceutical manufacturers , targets in our lawsuit-obsessed country. 2015)), Booker v. 604 (2011), and Mutual Pharmaceutical Co. It is fair to say that Bexis co-founded this blog (in 2006!) Federal preemption involving OTC drugs is the subject of today’s musings. 379r(a)(1).
The court dismissed the design-defect claim as pleaded, holding it impliedly preempted under Mutual Pharmaceutical Co. b)(2) not to make such a change without first obtaining FDAapproval.” Ortho-McNeil-Janssen Pharmaceuticals, Inc. , Ortho-McNeil-Janssen Pharmaceuticals, Inc. , Bartlett , 570 U.S. 3d 281 (6th Cir.
2015), Tersigni v. It quoted from Bartlett on impossibility preemption and the rejection of the argument that it “could be avoided because [the] pharmaceutical company could comply with seemingly irreconcilable state and federal laws by ceasing production of the drug altogether.” Wyeth , 619 F.3d 3d 632 (6th Cir. Wyeth , 85 A.3d
But in prescription medical product liability litigation, products must receive FDAapproval, clearance or other authorization (hereafter, collectively referred to as “approval” for short) before they can be marketed. to determine whether a proposed alternative drug would have received FDAapproval.” at 237-38.
Novartis Pharmaceuticals Corp. , Luitpold Pharmaceuticals, Inc. , Texas, unlike most states, enforces a strong statutory presumption that prescription medical product warnings complying with FDA requirements imposed by “pre-market approval or licensing of the product” are adequate as a matter of law. 2d 898, 909-10 (W.D.
2019), or Mutual Pharmaceutical Co. Plaintiffs Legal Committee , 531 U.S. Kent , 552 U.S. 440 (2008) Buckman was not cited at all in the Merck Sharp & Dohme Corp. Albrecht , 139 S. Bartlett , 570 U.S. 472 (2013), implied preemption decisions, cited only by the dissent in Wyeth v. Levine , 555 U.S. 555 (2009), and rated only a “ cf.
Bayer Healthcare Pharmaceuticals Inc. 2022 WL 17348351, at *4. “[F]or certain categories of drugs” – including this one – “the monograph system replaces the individualized NDA approval process with a rulemaking process.” Here’s a link to the official statement on the FDA’s website. 2022 WL 16951538 (S.D.N.Y.
June 24, 2022), we fully expect attempts by such states to ban FDA-approved prescription drugs that can be used to bring about abortions by chemical means. But when the FDA has approved a product, states no longer have the power to prohibit their sale or use for FDA-approved indications. 660, 664 (1962).
2012), addressed a challenge to the application of Idaho’s Pain-Capable Unborn Child Protection Act to criminalize the use of an FDA-approved abortifacient medication obtained through an internet prescription and mailed to the plaintiff from out of state. As we discussed here , McCormack v. Hiedeman, 694 F.3d 3d 1004 (9th Cir.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.














































Let's personalize your content