This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Livornese — I saw the sign…and the answer is no—FDA-approved labeling apparently is not enough under state failure-to-warn laws, according to certain courts. The GAO Report further explained that the agency did not have the resources to regulate the estimated 100,000 OTC drugs marketed through the monograph process.
Each of these issues, says Novartis, “independently renders the agency’s decision unlawful, and invalidates the agency’s approval of the MSN product.” While denying a Citizen Petition from Novartis asking FDA to refuse to approve any ANDA that omits the new dosing regimen or changes the indication, FDAapproved MSN’s ANDA on July 24, 2024.
To further facilitate generic drug development, and to assist the generic pharmaceutical industry in this process, the FDA publishes product-specific guidances (PSGs) describing the agency’s current thinking and expectations on how to develop generic drug products that are therapeutically equivalent to their brand name counterparts.
156, a patent may be extended only once (even if it would be eligible for extension on more than one occasion because it applies to several FDA-approved products), and only one patent may be extended for each regulatory review period. RE 44,638 and with respect to each NDA approval, See FDA Docket Nos.
The 51 regulations that FDA is currently working on The FDA today unveiled its much-anticipated Spring 2023 Unified Agenda, a document outlining the regulations the agency plans to release in 2023 and beyond. The anticipated date of publication is June 2023, meaning we should see this regulation imminently.
156, as added by the 1984 Hatch-Waxman Amendments, for certain FDA-regulated products. The use of a surrogate endpoint can considerably shorten the time required prior to receiving FDAapproval. Karst — Earlier this week, we posted Part 1 of our three-part series on U.S. Patent Nos.
Years of hard work, supported by the National Institutes of Health and the Cystic Fibrosis Foundation, painstakingly worked out the normal function of the protein that is altered in CF, called the cystic fibrosis transmembrane regulator (CFTR). Credit: Zhang & Chen, 2016, Cell 167, 1586–1597. About 30,000 Americans have CF.
This year we completed investigational new drug (IND) enabling studies of ONC-841, which is an antagonist of Siglec-10, and received US Food and Drug Administration (FDA) approval for first-in-human study (FIH) in patients with solid tumours. The first patient has been dosed in the last quarter.
The standard for the audit used depends on the type of vendor and would include the applicable regulations and ICH guidelines. The sponsor-initiated IND is conducted by a pharma company seeking commercial approval. The term drug substance and drug product are found in the NDA regulations 21 CFR 314.3, of this guidance.
In a new final rule, FDA carves out a regulatory niche for medical gases Industry has been lobbying FDA and Congress to regulate medical gases different from other types of drug products since the 1970s. Following this process, FDASIA directed the FDA to submit a report to Congress summarizing its findings.
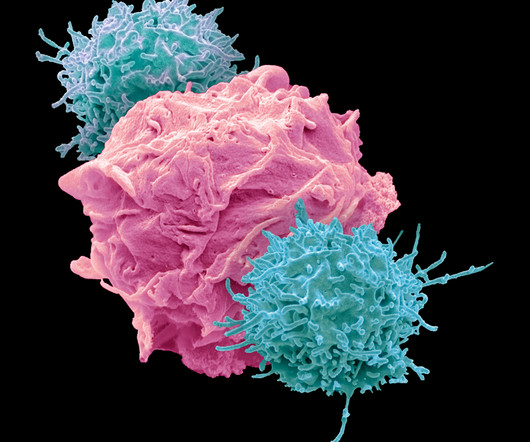
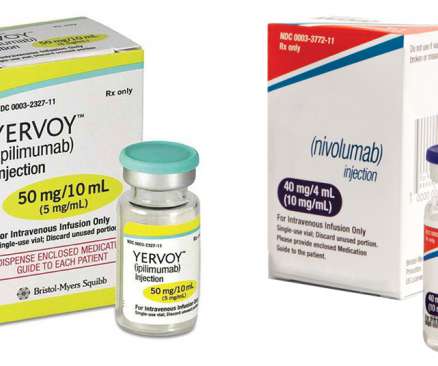
In July 2014, Opdivo was the first PD-1 immune checkpoint inhibitor to receive regulatory approval anywhere in the world. Opdivo is currently approved in more than 65 countries, including the United States, the European Union, Japan and China. CTLA-4 is a negative regulator of T-cell activity. About Yervoy.
In July 2014, Opdivo was the first PD-1 immune checkpoint inhibitor to receive regulatory approval anywhere in the world. Opdivo is currently approved in more than 65 countries, including the United States, the European Union, Japan and China. CTLA-4 is a negative regulator of T-cell activity. About Yervoy.
BY AMANDA CONTI SEP 13, 2023 1:58 PM CDT Quick background on nonprescription drug regulation Nonprescription drugs, also known as over-the-counter (OTC) drugs, are regulated differently than traditional prescription drugs. The FDA will follow these procedures for both agency-initiated operations (e.g.,
What We Expect the FDA to do in October 2023 In this ongoing feature, AgencyIQ looks at public data to determine what the FDA is likely to do in the month ahead, including key deadlines, meetings, events, planned regulations, comment periods and more.
What We Expect the FDA to do in November 2023 In this ongoing feature, AgencyIQ looks at public data to determine what the FDA is likely to do in the month ahead, including key deadlines, meetings, events, planned regulations, comment periods and more.
In proposed rule, FDA offers up a (surprisingly sparse) list of drugs that present demonstrable compounding difficulties FDA yesterday released a long-awaited proposed rule on a list of drug products that present demonstrable difficulties for compounding. The FDA sought input on these criteria from the PCAC in 2015 and again in 2016.
The recruitment period for the study spanned from December 2015 to July 2017. Food and Drug Administration (FDA) approval for nmCRPC on February 14, 2018 and was approved for mCSPC on September 17, 2019. ERLEADA ® received U.S. 2 To date, more than 10,000 patients worldwide have been treated with ERLEADA ®.
The cost of artificially short expiration dates: Worsened shortages, higher costs and more waste As drug shortages have made headlines over the past few years, the FDA has announced the extension of expiration dates on a variety of drug products. In most states, this defers to the USP recommendation.
Janssen has worked with BARDA since 2015 on innovative solutions for influenza, chemical, biological, radiation and nuclear threats and emerging infectious diseases such as Ebola. COV2-S (SARS-CoV-2 vaccine) FDAApproval History. Department of Health and Human Services (HHS). i] [link] [ii] The B.1.351 Posted: January 2021.
FDAapproval for adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to methotrexate. 2015 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. 3 In August 2019 , RINVOQ received U.S. Available at: [link]. Available at: [link].
Food and Drug Administration (FDA) approved the update of the Imbruvica Prescribing Information to include efficacy and safety data for the combination of Imbruvica with rituximab for the treatment of Waldenström’s macroglobulinemia (WM). and by AbbVie outside of the U.S. AbbVie announced that the U.S.
What We Expect the FDA to do in December 2023 In this ongoing feature, AgencyIQ looks at public data to determine what the FDA is likely to do in the month ahead, including key deadlines, meetings, events, planned regulations, comment periods and more.
Streamlining FDAApprovals : The FDA should work with drug regulators in other countries to create a common portal for submitting generic drug applications. This can streamline the approval process and increase the number of generic drugs available in the market. References Schulman, K. JAMA, 314(14), 14691470.
2 Along the way, they argue that political polarization around IVF has prevented federal-level regulation, resulting in a “ Wild West ” of reproductive technology — a state of affairs that has enabled some abuses (and ineffectual fertility treatments), but allows for greater innovative freedom overall.
As the DDL blog has previously reported , Michigan’s longstanding presumption of non-defectiveness applicable to FDA-approved drugs was recently repealed by the Michigan legislature in S.B. As this provision is newly applicable to FDA-approved products, it has not been tested by the courts on this issue. .
FDA litigation that is now before the Supreme Court. Congress created an FDAapproval process that is both rigorous and thorough, and pharmaceutical companies invest billions of dollars in research and development to meet FDA’s scientific standards. We were hardly alone. & Man’l Epid’y. Studnicki J, Harrison D.J.,
The plaintiff failed to identify any method by which a generic (or any other) drug manufacturer could add a Prop 65 warning without deviating from FDA-approved labeling, thereby violating federal law. Express and implied preemption operate independently ( see Buckman ), thus the express Prop 65 saving clause was irrelevant.
Constitution’s Supremacy Clause, it strikes a balance between state and federal power on issues of public health and safety, and raises questions about whether our country’s approach to regulation and litigation makes sense for pharmaceuticals and medical devices (spoiler alert: it doesn’t). 2015)), Booker v. 379r(a)(1). 379r(a)(1).
b)(2) not to make such a change without first obtaining FDAapproval.” As construed by the court, the complaint alleged that the biologic approved by the FDA is defective as a matter of state law. 2015), while as we discussed here , the Seventh Circuit reached the contrary conclusion in Kaiser v. 3d 281 (6th Cir.
2015), finally gave appellate recognition to the preemption of design defect claims for FDA-approved branded prescription drugs. FDAapproved the drug with its particular formulation and the manufacturer could not have changed the formulation on its own. As detailed here , the decision in Yates v. 3d 281 (6th Cir.
But in prescription medical product liability litigation, products must receive FDAapproval, clearance or other authorization (hereafter, collectively referred to as “approval” for short) before they can be marketed. to determine whether a proposed alternative drug would have received FDAapproval.” at 237-38.
280 (1995), decision that he joined in 1995 He joined a decision that found no preemption by virtue of a governmental decision not to regulate in Myrick and did not object to boilerplate description of obstacle preemption as an accepted preemption category Then in the 5-4 Geier v. Jones , 225 U.S. Davidowitz , 312 U.S. Davidowitz , 312 U.S.
The motions were teed up as to the claims of five plaintiffs whose use occurred solely during a period when it was uncontested that the generics had FDA-approved labeling that matched that of the branded drug.
Then, the decision makes a hash of the relevant administrative record, ignoring what the preemptive FDAregulations said in favor of material from the Federal Register that never actually made it into the regulations themselves. The FDA also promulgated an acetaminophen-specific OTC regulation. 201.326(a)(1).
The plaintiffs in Pfaff , who opted out of the settlement, had originally brought suit in California federal court back in 2015. Preemption turns on the availability of the FDA’s changes being accepted (“CBE”) regulation, 21 C.F.R. 12 (citation and quotation marks omitted).
After opportunity for notice and comment, the FDA promulgated literally hundreds of regulations classifying device types. Despite that classification, the statute allowed it to be cleared even before approval based on its equivalence to a device on the market in 1976, when MDA device regulation began. See 21 C.F.R.
June 24, 2022), we fully expect attempts by such states to ban FDA-approved prescription drugs that can be used to bring about abortions by chemical means. On occasion, states have attempted to prohibit sale and/or use of non-abortion-related FDA-approved products, but with one exception, these efforts have not resulted in litigation.
2012), addressed a challenge to the application of Idaho’s Pain-Capable Unborn Child Protection Act to criminalize the use of an FDA-approved abortifacient medication obtained through an internet prescription and mailed to the plaintiff from out of state. As we discussed here , McCormack v. Hiedeman, 694 F.3d 3d 1004 (9th Cir.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.









































Let's personalize your content