This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
This approval comes after GSK submitted results from its Phase 3 trial which enrolled 108 patients. Today’s approval gives these patients access to biologic treatment for the first time and demonstrates our commitment to maximising Nucala’s impact on eosinophil-driven diseases. “.
Additionally, mepolizumab was the first biologic therapy indicated for adults with eosinophilic granulomatosis with polyangiitis (EGPA) and also the first biologic to be approved for patients aged 12 years and older with hypereosinophilic syndrome (HES). With 41 clinical trials, mepolizumab has been studied in over 4,000 patients.
Food and Drug Administration (FDA) has approved PREVNAR 20 (Pneumococcal 20-valent Conjugate Vaccine) for the prevention of invasive disease and pneumonia caused by the 20 Streptococcus pneumoniae (pneumococcus) serotypes in the vaccine in adults ages 18 years and older. Following today’s FDAapproval, the U.S.
ONCR-177, its lead oncolytic herpes simplex virus (oHSV)-based immunotherapy, is in human trials as a monotherapy and in combination with Merck’s Keytruda (pembrolizumab) for a range of solid tumors. In addition to its lead program, which began Phase I testing in 2020, the company expects to nominate three clinical candidates in 2021.
Opdivo- based therapies have now demonstrated positive results in Phase 3 trials in earlier stages of four different types of cancer: non-small cell lung cancer, esophageal/gastroesophageal junction cancer, bladder cancer and melanoma. Surgery was rarely canceled due to adverse events, only affecting two patients in each arm of the trial.
Strikingly, a strong genetic link between target and disease biology is emerging as a predictor for success in clinical trials. Why Clinical Trials Stop: The Role of Genetics. Human genetics evidence supports two-thirds of the 2021 FDA-approved drugs. Are drug targets with genetic support twice as likely to be approved?
This year we completed investigational new drug (IND) enabling studies of ONC-841, which is an antagonist of Siglec-10, and received US Food and Drug Administration (FDA) approval for first-in-human study (FIH) in patients with solid tumours. Biomarkers are the holy grail of clinical trials.
The use of a surrogate endpoint can considerably shorten the time required prior to receiving FDAapproval. If the confirmatory trial shows that the drug actually provides a clinical benefit, then the FDA grants traditional approval for the drug. That did not happen. Patent Nos.
Applications based on positive results from the Phase 3 CheckMate -648 trial, in which both Opdivo -based combinations demonstrated a significant survival benefit over chemotherapy alone. Bristol Myers Squibb thanks the patients and investigators involved in the CheckMate -648 trial. About CheckMate -648.
The Breakthrough Therapy designation aims to expedite the development and review of drugs that are intended to treat a serious condition when preliminary clinical evidence indicates that the drug may demonstrate substantial improvement on a clinically significant endpoint(s) over already available therapies that have received full FDAapproval.
In early August, risdiplam, an orally available small molecule that binds to a stem-loop structure in the SMN2 pre-mRNA, received FDAapproval for the treatment of spinal muscular atrophy , a devastating genetic disease. The agent, onasemnogene abeparvovec, was approved by the FDA in 2019 and is marketed as Zolgensma.
Approval based on Phase 2 CheckMate -142 trial results showing nearly two-thirds of patients responded to the Opdivo plus Yervoy combination with durable responses observed. Opdivo plus Yervoy is the first dual immunotherapy regimen approved in the European Union for colorectal cancer. CheckMate -142 Efficacy and Safety Results.
.–( BUSINESS WIRE )– Bristol Myers Squibb (NYSE: BMY) today announced the Phase 3 CheckMate -577 trial evaluating Opdivo (nivolumab) as an adjuvant therapy for patients with resected esophageal or gastroesophageal junction (GEJ) cancer met its primary endpoint of disease-free survival (DFS) at a pre-specified interim analysis.
The study is an open-label, exploratory phase I/II trial adding ONCOS-102 to SoC chemotherapy (pemetrexed/cisplatin) in first- and second- (and later) line treatment of MPM to assess safety, immune activation and efficacy versus SoC only. months (Baas 2020), and this is expected to serve as a benchmark for further approvals. .”
Helping to fuel this optimism are preliminary results released this week from early-stage clinical trials of three next-generation, triple combination therapies that modulate the function of CFTR. According to preliminary data from one Phase 1 and two Phase 2 clinical trials reported by Vertex Pharmaceuticals Inc.,
The safety profiles of Opdivo and chemotherapy in this trial are reflective of the known safety profiles of Opdivo and chemotherapy in first-line gastric and esophageal cancers. The company remains blinded to data from this arm and the trial continues in follow-up to allow the data to mature. PRINCETON, N.J.–(
The sponsor is the pharmaceutical company conducting the trial. If you mean using a different contract research organization (CRO) for the different phases of clinical trials – that’s different. Also consider CRO oversight, trial management, data handling and record keeping, as well as allocation of responsibilities.
January 29, 2021 – Johnson & Johnson (NYSE: JNJ) (the Company) today announced topline efficacy and safety data from the Phase 3 ENSEMBLE clinical trial, demonstrating that the investigational single-dose COVID-19 vaccine in development at its Janssen Pharmaceutical Companies met all primary and key secondary endpoints.
FDA’s Vaccines and Related Biological Products Advisory Committee (VRBPAC) on February 26, 2021. “We We are thankful for the efforts of all those who have volunteered to participate in our clinical trials, our scientists, collaborators, clinical trial sites and investigators. There is no FDA-approved vaccine to prevent COVID-19.
CheckMate -743 remains the only Phase 3 trial to show a survival benefit with first-line immunotherapy treatment in patients with MPM. First-line nivolumab + cabozantinib vs sunitinib in patients (pts) with advanced renal cell carcinoma (aRCC) in subgroups based on prior nephrectomy in the CheckMate 9ER trial. Dr. Camillo Porta.
Until recently, the FDA relied on a monograph process through which firms could bring OTC drugs to market without FDAapproval so long as it adhered to pre-set terms under the monograph. Finally, the committee came up with a list of recommendations for the structure of additional trials, listed in the meeting minutes.
In 2017, Opdivo was granted accelerated approval by the FDA as a single agent for patients with HCC who have been previously treated with sorafenib. The accelerated approval was based on tumor responses from the Phase 1/2 CheckMate -040 trial.
There was nothing left over for human trials, until an opportune call came from Elon Musk’s secretary. The money was earmarked for human trials and, afterward, to develop a neural prosthesis that could augment memories in healthy adults. ” Musk later wrote a check for one million dollars. That had to help!
Similarly, only one product in 2023 was recommended for approval under “ exceptional circumstances ” – Loargys. This approval type has always been uncommon – before the pandemic, this pathway was used just eight times between 2015 and 2019. [ See AgencyIQ’s analysis of this approval type from 2015-2021 here.]
While stimulant use disorder is increasing, there are currently no FDA-approved medications. According to the Substance Abuse and Mental Health Services Administration (SAMHSA), “between 2008 and 2015, amphetamine-related hospitalizations more than tripled, increasing from 55,447 instances to 206,180.
The recruitment period for the study spanned from December 2015 to July 2017. Food and Drug Administration (FDA) approval for nmCRPC on February 14, 2018 and was approved for mCSPC on September 17, 2019. 6] ERLEADA ® is being studied in five Phase 3 registrational clinical trials. ERLEADA ® received U.S.
2,4 This analysis included data pooled from six rheumatoid arthritis Phase 3 clinical trials and included more than 3,000 patients with over 7,000 PY of exposure of RINVOQ 15 mg, as well as data of HUMIRA and MTX. More information on this trial can be found at www.clinicaltrials.gov (NCT02629159). The approved dose for RINVOQ is 15 mg.
billion increase in the Skyrizi contingent consideration liability due to higher estimated future sales driven by stronger market share uptake and favorable clinical trial results as well as lower interest rates. Recorded a $4.7 and by AbbVie outside of the U.S. Imbruvica is jointly developed and commercialized with Janssen Biotech, Inc.
The proposed rule will identify certain bulk drug substances that FDA has considered and is proposing to place on the 503A Bulks List and certain bulk drug substances that FDA has considered and is proposing not to include on the 503A Bulks List.
12/29/2023 FDORA, Section 3602 Clinical Trials Modernization : FDA is directed to require the submission of a “diversity action plan” for all Phase 3 clinical trials of new drugs. FDA is directed to issue new draft guidance or update existing guidance regarding Diversity Action Plans for clinical studies.
Such a situation is commonplace in the clinical trial realm, in which investigational drug products which are not already FDAapproved are administered to patients. Under the Pandemic and All-Hazards Preparedness Reauthorization Act of 2013 (PAHPRA) , the FDA also has some authority to extend MCM expiration dates.
1] Zavegepant is an antagonist of the calcitonin gene-related peptide (CGRP) receptor currently in phase 3 trials in an intranasal formulation for the treatment of migraine. The clinical trial is expected to complete in September 2020. Zavegepant is indicated for the acute treatment of migraine with or without aura in adults. [1]
12/29/2023 FDORA, Section 3602 Clinical Trials Modernization : FDA is directed to require the submission of a “diversity action plan” for all Phase 3 clinical trials of new drugs. FDA is directed to issue new draft guidance or update existing guidance regarding Diversity Action Plans for clinical studies.
Food and Drug Administration (FDA) approved Inmazeb (atoltivimab, maftivimab and odesivimab-ebgn) for the treatment of infection caused by Zaire ebolavirus in adult and pediatric patients, including newborns of mothers who have tested positive for the infection. TARRYTOWN, N.Y., NASDAQ: REGN) announced today that the U.S.
Metabolism of 2024 FDAapproved small molecules part 1 By Julia Shanu-Wilson Involvement of active metabolites The number of small molecules approved by the FDA in 2024 totaled 31 out of 50 NMEs with 56% (28) of these comprising small molecule therapies. 2015 May;54(5):457-71. What’s next? Clin Pharmacokinet.
7] Treosulfan was authorized for medical use in the European Union in June 2019, [9] and approved for medical use in the United States in January 2025. [7] 14/L Trial II (NCT00822393), a randomized active-controlled trial comparing treosulfan to busulfan with fludarabine as a preparative regimen for allogeneic transplantation.
They ran extensive trials and experiments to inoculate people against this perennial killer. The 2014-2015 seasonal vaccine, for instance, was only 19 percent effective due to a mismatch between the selected strain and the circulating one. It’s still in preclinical development, but the hope is to move into clinical trials soon.
Food and Drug Administration (FDA) approved Inmazeb, a three-antibody cocktail to treat Ebola infections in adults and children. It has also been approved for newborns of mothers who have tested positive for Ebola. In 2019, the FDAapproved an Ebola vaccine, Merck’s Ervebo. “We Today it announced that the U.S.
After six years of such trials, Rock marveled as fertilized eggs formed zygotes and entered the cleavage stage of embryonic development. Various techniques for using abnormal sperm in fertilization were trialed through the late 1980s, but by 1995, the clear winner was intracytoplasmic sperm injection (ICSI). Since IVF in the U.S.
In particular, plaintiffs trying to force clinical trial sponsors to continue providing free drug after the end of the trial—something clinical trial participants are told is not a guaranty—have floundered. We do not even need to call out the emphasis that plaintiff lawyers place on FDAapproval when it suits them.
2023) (remote trial testimony cannot be compelled beyond Rule 45’s 100-mile limit on subpoenas) ( here ); Carson v. The FDA requires real science for warnings; thus it had not mandated any warning remotely resembling Prop 65. Bonta , 85 F.4th 4th 1263 (9th Cir. 4th 1030 (9th Cir. Monsanto Co. , 4th 1261 (11th Cir. Ethicon, Inc. ,
In two of these cases, our client won summary judgment at the trial court level and an appellate court ended up creating a new cause of action to accommodate the plaintiff’s theory (and lack of helpful testimony from the prescribing physician). 2015), Tersigni v. Wyeth , 619 F.3d 3d 632 (6th Cir. Wyeth , 85 A.3d 3d 434 (Pa.
But in prescription medical product liability litigation, products must receive FDAapproval, clearance or other authorization (hereafter, collectively referred to as “approval” for short) before they can be marketed. to determine whether a proposed alternative drug would have received FDAapproval.” at 237-38.
Maretta , 569 U.S. 483 (2013), concurrence regarding preemption concerning insurance policies of federal employees – supports preemption when the “ordinary meaning” of a federal statute “effectively repeals contrary state law” by virtue of a “direct conflict” Oneok, Inc. Learjet, Inc. , Albrecht , 139 S.Ct.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.








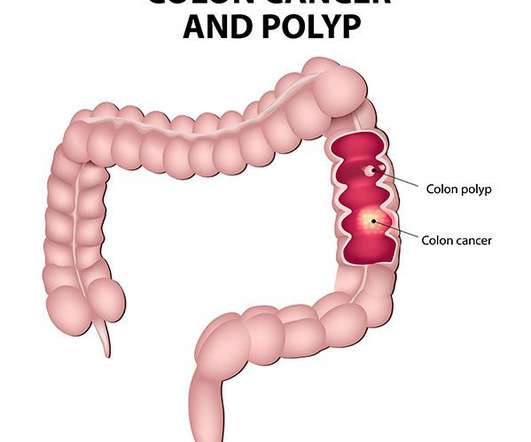




















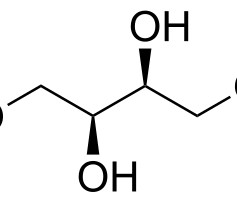












Let's personalize your content