This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
FDA conducted the eight-factor scheduling analysis required by the CSA in 2016 and found that marijuana continued to meet the scheduling criteria for remaining in schedule I. 12, 2016); Denial of Petition to Initiate Proceedings to Reschedule Marijuana, 81 Fed. 53,688 (Aug. 53,767 (Aug. 21 U.S.C. § at 53,700; 81 Fed.
Rescheduling out of schedule I would allow for the medical use of FDA-approved prescription drugs dispensed by DEA-registered, state licensed pharmacies pursuant to prescriptions issued by similarly DEA-registered, state licensed practitioners. Denial of Petition To Initiate Proceedings to Reschedule Marijuana, 81 Fed.
Food and Drug Administration (FDA) has approved PREVNAR 20 (Pneumococcal 20-valent Conjugate Vaccine) for the prevention of invasive disease and pneumonia caused by the 20 Streptococcus pneumoniae (pneumococcus) serotypes in the vaccine in adults ages 18 years and older. Following today’s FDAapproval, the U.S.
Scott and colleagues focused on six genes that encode potential drug targets licensed or in development by GlaxoSmithKline for the treatment of obesity or diabetes. That’s critical considering that just 1 in 10 drug candidates entering human clinical trials successfully goes on to receive FDAapproval [5]. 2016 March 4. [5]
The use of a surrogate endpoint can considerably shorten the time required prior to receiving FDAapproval. If the confirmatory trial shows that the drug actually provides a clinical benefit, then the FDA grants traditional approval for the drug. 8,128,929 was submitted on December 8, 2016 (Docket No.
Relevant here, FDA interpreted in Guidance that a proposed injectable biosimilar must “demonstrate that its product has the same strength as the reference product by demonstrating that both products have the same total content of drug substance (in mass or units of activity) and the same concentration of drug substance.”
If approved, Dupixent will be the first biologic medicine available in the U.S. In 2016, the FDA granted Breakthrough Therapy designation for Dupixent for the treatment of severe atopic dermatitis (in children aged 6 months to 11 years). to treat uncontrolled moderate-to-severe atopic dermatitis for these young children.
FDA addressed the expanding practice of drug compounding in 1992 by issuing a compliance policy guide that clarified that pharmacies which compounded products at certain scales, for certain purposes, or without FDAapproval were clearly operating “outside the bounds of traditional pharmacy practice.”
Under the terms of the amended agreement , originally executed in 2016 with Bristol Myers’ Celgene, an undisclosed fee was paid to Anokion to include KAN-101 for the treatment of celiac disease in the exclusive global collaboration.
The meeting took place before the launch of Neuralink, in July 2016. DARPA invested more than $200 million in support of the White House’s Brain Research through Advancing Innovative Neurotechnologies ( BRAIN ) Initiative between 2014 and 2016 alone. The company was not publicly unveiled until the following year.)
An update on the Medical Device Quality Management System Regulation : In February 2022, the FDA proposed a new rule which would effectively transition the agency away from its longstanding Quality System Regulation (QSR) in favor of the ISO 13485:2016 standard. Read our analysis of that rule here and here. ]
and Annex 1 Conference Joel Welch December 18 RAPS RAPS Webcast: FDA Forecast: What’s Next for the FDA in 2024 AgencyIQ Speakers December 21 HL7 REMS Public Call PDUFA Dates expected in November and December PDUFA dates represent the expected date of a regulatory decision by the FDA on a New Drug Application or Biologics License Application.
We wondered how given that HHS and the Drug Enforcement Administration (“DEA”) conducted eight-factor scheduling analyses in 2016, concluding that there was “no substantial evidence that marijuana should be removed from Schedule I.” 12, 2016) ; Denial of Petition to Initiate Proceedings to reschedule Marijuana, 81 Fed. 12, 2016).
Cannabis in schedule III would require a prescription issued by a DEA-registered, state-licensed practitioner. DEA and HHS last considered rescheduling cannabis in 2016. 12, 2016); Denial of Petition to Initiate Proceedings to Reschedule Marijuana, 81 Fed. 53,688 (Aug. 53,688 (Aug. 53,767 (Aug. 21 U.S.C. § at 53,689, 53,768.
December 2023 Medical Devices; Quality System Regulation Amendments (Final Rule) FDA intends to harmonize and modernize the Quality System regulation for medical devices. The revisions will update the existing requirements with the specifications of an international consensus standard for medical device manufacturers, ISO 13485:2016.
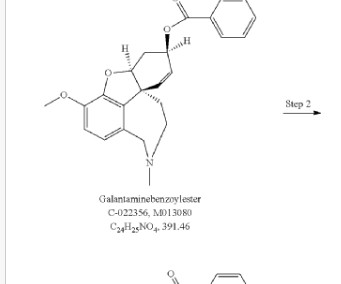
1] Society and culture Legal status Benzgalantamine was approved for medical use in the United States in July 2024. [1] Jump up to: a b c [link] ^ “Alpha Cognition’s Oral Therapy Zunveyl Receives FDAApproval to Treat Alzheimer’s Disease” (Press release). 2016 Jan 20;2(1):13-22. 8 August 2024.
FDA/HHS Analysis, 2023 FDA/HHS last conducted an eight-factor analysis of marijuana in 2016 and concluded that marijuana continued to meet schedule I criteria. 12, 2016); Denial of Petition to Initiate Proceedings to Reschedule Marijuana, 81 Fed. Denial of Petition to Initiate Proceedings to Reschedule Marijuana, 81 Fed.
There has been no evidence that marijuana’s schedule should change since the last rescheduling review in 2016. FDA has not approved marijuana for medical use because no double-blind, published studies show safety and efficacy for raw marijuana. In other words, no rescheduling.
The Complaint further alleges that data collected and submitted in support of full (as opposed to emergency use) FDAapproval demonstrated the misleading nature of earlier statements. The FDA, however, did not and does not share that belief. FDA (8/23/21) press release (emphasis original). 115, 125 (2016).
The FDA requires real science for warnings; thus it had not mandated any warning remotely resembling Prop 65. The plaintiff failed to identify any method by which a generic (or any other) drug manufacturer could add a Prop 65 warning without deviating from FDA-approved labeling, thereby violating federal law. 13) McGee v.
For instance, Sorsaia primarily involves a challenge to a state trying to prevent in-state use of an FDA-approved drug for its FDA-approved use. 115 (2016), eight years after Levine , there is no such presumption in express preemption either. Last week, the district court in GenBioPro, Inc. emphasis in original).
The core premise of Bexis’ article is very simple: Once the FDA has said “yes” and approved a particular drug for a particular indication (“intended use”) for sale in the United States, federal preemption precludes any state from saying say “no” and trying to ban that same FDA-approved drug. In GenBioPro, Inc.
Texas, unlike most states, enforces a strong statutory presumption that prescription medical product warnings complying with FDA requirements imposed by “pre-market approval or licensing of the product” are adequate as a matter of law. 2016) (applying the Texas warning presumption to a generic drug eluting patch).
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.





























Let's personalize your content