This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The 2018 Farm Bill. The 2018 Farm Bill removed hemp from the Controlled Substance Act. Importantly, the 2018 Farm Bill preserved FDA authority to regulate products with cannabis or cannabis-derived compounds under the Federal Food, Drug, and Cosmetic (FD&C) Act and Section 351 of the Public Health Service Act.
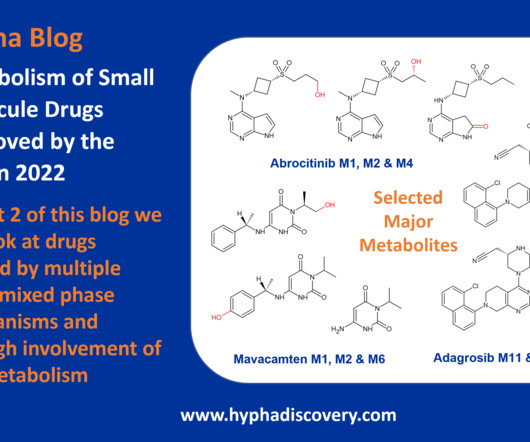
Metabolism of 2022 FDAapproved small molecule drugs part 2 Mixing it Up By Julia Shanu-Wilson In Part 1 of this topic we looked at metabolism of the small molecule drugs approved by the FDA in 2022 that were mediated by CYP3A4. link] [2] Zhang and Tang, 2018. 2018 Sep;8(5):721-732. Br J Pharmacol.
As with other FDA-regulated products, such as human drugs and medical devices, the “regulatory review period” is composed of a “testing phase” and a “review phase.” The “review phase” is the period between the initial submission and approval of the NADA. FDA’s PTE regulations at 21 C.F.R.
156, a patent may be extended only once (even if it would be eligible for extension on more than one occasion because it applies to several FDA-approved products), and only one patent may be extended for each regulatory review period. 2018) , Arnold Partnership v. Part 1: Multiple PTEs Under the PTE statute at 35 U.S.C. §
Interneuron migration is regulated by L-type calcium channels (LTCCs), encoded by the CACNA1C gene. Recently, a network-based drug-screening platform has been developed to test FDA-approved drugs for Alzheimer’s disease (AD) using 1,300 iPSC-derived organoids from 11 sporadic AD patients. 12(3) (2023). link] Madhavan M, et al.
In 2018, the FDA published a detailed guidance on labeling requirements for biosimilar products. However, the 2018 guidance it did not offer advice for interchangeable product labeling. The FDA published recommendations for interchangeable products separately in a completely new guidance , issued two years later.
BY CHELSEY MCINTYRE, PHARMD | JUN 3, 2024 8:43 PM CDT Regulatory background: DSHEA and dietary supplements The Dietary Supplement Health and Education Act ( DSHEA ) of 1994 defines the FDA’s authority in the regulation of dietary supplement products and dietary ingredients. See AgencyIQ’s analysis of the draft guidance here. ]
New Phase III data from SAkuraStar and SAkuraSky studies demonstrate reduced severity of relapses with ENSPRYNG (satralizumab), recently FDA-approved as the first and only subcutaneous treatment for adults living with anti-aquaporin-4 (AQP4) antibody positive neuromyelitis optica spectrum disorder (NMOSD).
Food and Drug Administration (FDA) approved EVRYSDI for the treatment of SMA in adults and children 2 months of age and older. EVRYSDI was granted PRIME designation by the European Medicines Agency (EMA) in 2018 and Orphan Drug Designation by FDA and EMA in 2017 and 2019, respectively. Europe and Japan.
BY AMANDA CONTI SEP 13, 2023 1:58 PM CDT Quick background on nonprescription drug regulation Nonprescription drugs, also known as over-the-counter (OTC) drugs, are regulated differently than traditional prescription drugs. The FDA will follow these procedures for both agency-initiated operations (e.g.,
Food and Drug Administration (FDA) approved the update of the Imbruvica Prescribing Information to include efficacy and safety data for the combination of Imbruvica with rituximab for the treatment of Waldenström’s macroglobulinemia (WM). and by AbbVie outside of the U.S. AbbVie announced that the U.S.
First and only FDA-approved subcutaneous treatment option for anti-aquaporin-4 antibody positive NMOSD that can be self-administered by a person with NMOSD or a caregiver every four weeks. First and only approved therapy for NMOSD designed to target and inhibit interleukin-6 receptor activity, using novel recycling antibody technology.
Food and Drug Administration (FDA) approved Pfizer Inc.’s LORBRENA is now indicated for adults with metastatic NSCLC whose tumors are ALK-positive as detected by an FDA-approved test. LORBRENA is now indicated for adults with metastatic NSCLC whose tumors are ALK-positive as detected by an FDA-approved test.
ENSPRYNG is the first and only FDA-approved subcutaneous, self-administered medicine for NMOSD and the first medicine for NMOSD that is designed to target the interleukin-6 receptor, which is believed to play a key role in the inflammation associated with this disorder.”. ENSPRYNG is approved in Canada, Japan, Switzerland and the U.S.
Following FDAapproval, these products are granted seven years of marketing exclusivity, preventing FDA from approving the “same drug for the same disease or condition” if the applicant does not have the right to reference the product (i.e., DORIS MATSUI (D-Calif.) and GUS BILIRAKIS (R-Fla.), TAMMY BALDWIN (D-Wisc.).
The cost of artificially short expiration dates: Worsened shortages, higher costs and more waste As drug shortages have made headlines over the past few years, the FDA has announced the extension of expiration dates on a variety of drug products.
” The pivotal Phase 3 RESONATE-2 study served as the basis for the FDAapproval of IMBRUVICA as a single-agent in first-line treatment for CLL/SLL in 2016, following initial approval for relapsed/refractory (R/R) patients in 2014 based on the RESONATE study. IMBRUVICA ® is the only FDA-approved medicine in WM and cGVHD.
According to the World Health Organization, esophageal cancer is currently the sixth most common cause of cancer death in the world and is estimated to have caused over 508,000 deaths in 2018. Persons”, as such term is defined in Regulations under the U.S.
” Interchangeable products can be substituted for the reference product at the pharmacy level, without prescriber intervention, within state and local regulations. In the last decade, the FDA has held various meetings and published several guidance documents to provide development recommendations to sponsors of biosimilar products.
For more than a century, scientists thought of gas vesicles as little more than a natural curiosity and, later, as a way for microbes to regulate buoyancy. A major breakthrough came in January 2018, when a paper in Nature indicated, for the first time, that gas vesicles could be expressed in E. coli cells.
BY RACHEL COE, MSC JUL 18, 2023 11:14 PM CDT Oncology endpoints: The growing pains of an evolving field FDAapproval of a drug or biologic relies on whether there is “substantial evidence” of safety and effectiveness derived from “adequate and well-controlled clinical investigations” according to the Federal Food, Drug, and Cosmetic (FD&C) Act.
Securities and Exchange Commission Regulation G. REDUCE-IT, conducted over seven years and completed in 2018, followed 8,179 patients at over 400 clinical sites in 11 countries with the largest number of sites located within the United States. REDUCE-IT was conducted based on a special protocol assessment agreement with FDA.
In 1940, Charles Drew—the first African-American researcher to earn a doctor of medical science degree at Columbia University—developed an ingenious method for separating and storing plasma , the liquid part of blood that contains essential proteins that promote clotting and regulate blood pressure. Science Robotics (2018).
In 2022, the FDAapproved the first LBP – Rebyota (RBX2660; Ferring Pharmaceuticals) – but has yet to approve a recombinant LBP. The regulation of dietary supplements in the U.S. is retroactive, as opposed to the proactive drug approval pathway. Some probiotics are also sold as plain old conventional foods.
Food and Drug Administration (FDA) approval for nmCRPC on February 14, 2018 and was approved for mCSPC on September 17, 2019. ASCO Answers: Prostate Cancer (2018). ERLEADA ® is an androgen receptor (AR) inhibitor indicated for the treatment of patients with nmCRPC and for the treatment of patients with mCSPC.
Food and Drug Administration (FDA) approval for nmCRPC on February 14, 2018 and was approved for mCSPC on September 17, 2019. FDA on April 28, 2011 and by the European Commission on September 7, 2011. FDA on February 8, 2018. [vi] vi] Since its first approval in the U.S.
FDAapproval for adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to methotrexate. Nat Rev Dis Primers 4, 1(2018). 14 In August 2019 , RINVOQ received U.S. Atopic Dermatitis: Global Epidemiology and Risk Factors. Ann Nutr Metab 2015;66(suppl 1):8–16.
Food and Drug Administration (FDA) approved the supplemental New Drug Application (sNDA) for Lorbrena, expanding the indication to include first-line treatment of people with anaplastic lymphoma kinase (ALK)-positive non-small cell lung cancer (NSCLC). The FDA action converts the 2018 accelerated approval to full approval.
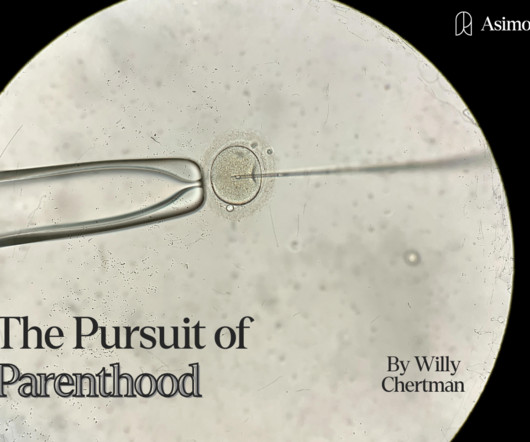
2 Along the way, they argue that political polarization around IVF has prevented federal-level regulation, resulting in a “ Wild West ” of reproductive technology — a state of affairs that has enabled some abuses (and ineffectual fertility treatments), but allows for greater innovative freedom overall.
Allegations that FDA-approved drug warnings are inadequate justify taking judicial notice of those warnings on a motion to dismiss. What such agency approved warnings say “can be accurately and readily determined from sources whose accuracy cannot reasonably be questioned.” quoting Fed.
When you hear Class III medical device product liability case, you should look for all claims to be dismissed unless there is something as unusual as a basis to claiming the plaintiff’s particular device deviated from its FDA-approved specifications. An unfortunate fiction developed post- Buckman —particularly after Riegel v.
Therefore, the warnings that were approved to accompany the drug included the risks of vaginal bleeding and endometrial changes. Plaintiff was prescribed the drug in 2018 and used it for approximately 10 months. Plaintiff also premised her failure to warn claims on a failure to report adverse events to the FDA.
June 24, 2022), we fully expect attempts by such states to ban FDA-approved prescription drugs that can be used to bring about abortions by chemical means. On occasion, states have attempted to prohibit sale and/or use of non-abortion-related FDA-approved products, but with one exception, these efforts have not resulted in litigation.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.


































Let's personalize your content