This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The Prescription Drug User Fee Act (PDUFA) has been a cornerstone of the U.S. Food and Drug Administrations (FDA) drug approval process since its inception in 1992. With the implementation of PDUFA VII (2023-2027), user fees for FY 2025 have been published, reflecting a notable increase that will impact pharmaceutical []
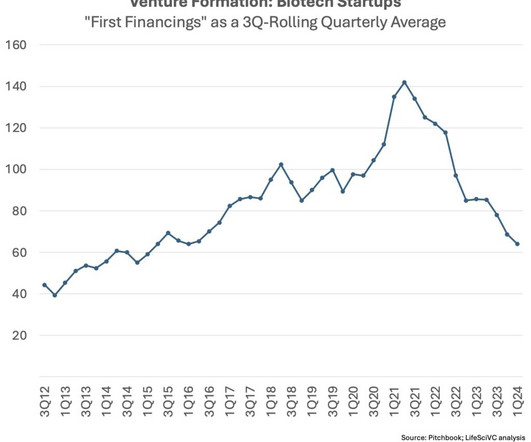
Further, the competitive intensity in certain areas makes the “clinical do-ability” too challenging, even if those drugs could likely be beneficial. Fewer startups over time should ameliorate this crowding dynamic to some extent.
In the ever-evolving field of medical science, artificial intelligence (AI) has emerged as a revolutionary tool, particularly in drug discovery. As a newer alternative to the time-consuming nature of traditional candidate screening and drug discovery, AI-designed drugs have started to make their mark, promising quick and efficient results.
This FR Notice and draft strategy document are part of FDA’s commitment under the Prescription Drug User Fee Act (PDUFA) Reauthorization Performance Goals and Procedures for Fiscal Years 2023-2027 (PDUFA VII), wherein FDA committed to advance the use and implementation of innovative manufacturing.
This blog provides an update on the DHT-related PDUFA VII goals that were targeted for completion in the first two quarters of FDA’s Fiscal Year (FY) 2023, including: By the end of Q2 FY 2023, FDA will establish a DHT framework document to guide the use of DHT-derived data in regulatory decision-makings for drugs and biological products.
Soon after, the Agency announced beginning on March 27, 2023, the FDA generic drug program would resume in-person FTF meetings, but caveated that the in-person FTF meeting option would only be available for pre-ANDA product development meetings and pre-submission meetings for which the applicant requests this in-person FTF meeting format.
Ultimately, this module will interact with CTIS , the drug clinical trial information system. The independent auditing schedule showed publication of full functionality in mid-2027. Another notable challenge has been development of the clinical investigation and performance study module (CI/PS).
Taking a step back, a Suitability Petition is used when an ANDA applicant wants to submit an ANDA that differs from its Reference Listed Drug in strength, dosage form, route of administration, or, in the case of a combination drug, active ingredient. Under 21 C.F.R.
These products are devices as defined in section 201(h) (1) of the Federal Food, Drug, and Cosmetic Act (the act), and may also be biological products subject to section 351 of the Public Health Service Act, including when the manufacturer of these products is a laboratory.
The FDA introduced the Type D Meeting on October 1, 2022, as part of the Prescription Drug User Fee Act (PDUFA) VII Commitment Letter. Prescription Drug User Fee Act (PDUFA) VII The Prescription Drug User Fee Act (PDUFA VII) is a law that was signed and took effect on September 30, 2022.
Food and Drug Administration’s Office of Orphan Products Development has granted Orphan Drug Designation to Uttroside-B , a small molecule chemotherapeutic for the treatment of hepatocellular carcinoma (HCC), the most common form of liver cancer. billion by 2027.
NEW YORK , Jan.
Q BioMed Inc.
It owned the societal name of the Canadian Association for Research on Drug Safety. As experts in early-phase drug development , from discovery through Phase II clinical research , we at Altasciences can share with STC members our accumulated knowledge and experience, and provide mentorship for future toxicologists.
How have pre-submission meetings for generic drug applicants changed under GDUFA III? Under GDUFA III, the scope and purpose of pre-submission meetings for generic drug developers has changed. One of the core goals of the GDUFA program has been to increase the efficiency of the generic drug review program.
Koblitz — Neither Jazz Pharmaceuticals nor Avadel CNS Pharmaceuticals has taken the battle of sodium oxybate—a drug approved to treat narcolepsy—lying down. Like in the multitude of Orphan Drug Act cases preceding this one, Jazz alleges that FDA’s authority under the Orphan Drug Act was limited by the statutory language.
By Faraz Siddiqui — Last Friday, the Delaware District Court rejected AstraZeneca’s lawsuit against the Medicare Drug Price Negotiation Program enacted under the Inflation Reduction Act (IRA) and CMS’s guidance implementing it. Opinion at 17. contradicts the plain text of the statute and therefore must be set aside.” . ; see also 42 U.S.C.
Kirschenbaum — The Inflation Reduction Act’s price negotiation program for drugs covered under Medicare Parts B and D (“Negotiation Program”) has been challenged in federal court. to transfer their patented pharmaceutical products to Medicare beneficiaries, for public use” at a government-dictated price that is a fraction of the drug’s value.
Class A devices needed to be IVDR-ready on the original date (May 26, 2022), but class D had until May 2025, class C until May 2026, and class B and sterile class A until May 2027. In-house tests (in the U.S. called laboratory-developed tests manufactured and used in a single lab) had until May 26, 2024.
Contract research organizations (CROs) are an integral partner of the drug development process, as they play a pivotal role supporting clinical trial conduct for pharmaceutical, biotechnology, and medical device sponsor companies. A rise of clinical trial outsourcing due to cost management, globalization, and digitalization.
FDA Warning Letters are often sent to manufacturers of generic drugs. Such products are often made in foreign facilities by staff less experienced with FDA regulatory requirements and at margins which may preclude significant investments in drug quality.
This activity could increase (or reduce the size of any decrease in) the market price of the Company’s common stock, the notes or the Company’s 2.50% Convertible Senior Notes due 2027 at that time. About BridgeBio.
Food and Drug Administration (FDA) made public a potentially game-changing proposal concerning the regulatory framework for laboratory-developed tests (LDTs). years after publication; no earlier than October 1, 2027) End of Enforcement Discretion for: Premarket review for high-risk IVDs. On September 29, 2023, the U.S. Phase 4 (3.5
There are also continued questions about regulatory capacity and what the field will look like going forward, with implications for drug developers who rely on diagnostic products. A sea change for lab tests, the implications for drug developers and outstanding questions about endpoints In the U.S.,
SEP 11, 2023 10:49 PM CDT Regulatory background Cosmetic ingredients in California are governed by the Federal Food, Drug & Cosmetic Act (FD&C Act), which provides that cosmetics produced or distributed for retail sale to consumers for their personal care are required to bear an ingredient declaration ( 21 CFR 701.3 ).
The regulation is focused on shifting the plastic pollution burden from consumers to producers, and requires the PRO to remit $500 million to the state each year between 2027 and 2037 for the California Plastic Pollution Mitigation Fund (totaling $5 billion).
In 2022, the MDCG offered a guidance document on the borderline between drugs and devices, and the manual itself containing decisions on products came out in September 2022. We’ve seen what’s been going on with the MDR, but the main concern we’ve heard of IVDR is the performance study application process is affecting drug trials.
In 2003, the CTD became the mandatory format for new drug applications in the EU and Japan, and the “strongly recommended format” for submission of applications to the FDA. The format and organization of the CTD is outlined in the ICH M4 Guideline.
October 1, 2027) in order to align with the next iteration of the device user fee program, the QS compliance requirement for LDTs could theoretically be in place by FY2027. The system as proposed could have compounding implications across the life sciences industry, including for drug development.
Can you tell us more about ZW191 and its potential as an FRα-targeting antibody-drug conjugate? ZW191 is comprised of a novel fully humanised IgG1 antibody covalently conjugated to a novel topoisomerase 1 inhibitor ZD06519, a camptothecin derivative, via endogenous interchain cysteines with a drug-to-antibody ratio (DAR) of eight.
In addition to reauthorizing for an additional five fiscal years—Fiscal Years 2023-2027—several drug, biological product, and medical device user fee provisions that were scheduled to sunset on September 30, 2022, FUFRA reauthorizes—but only through December 16, 2022—several other statutory provisions that were scheduled to expire.
This is about reducing the time and cost and increasing the predictability of going from concept to commercialization.” – Dr. Jeff Shuren During a presentation at the May 17, 2023 Food and Drug Law Institute (FDLI) Annual Conference, CDRH Director, Dr. Jeff Shuren discussed TAP with enthusiasm.
A January 2022 Seminars in Arthritis and Rheumatism publication noted that a “generic [drug] may only cost $1 million to $4 million and take two years to develop vs $100 million to $250 million and seven to eight years for a biosimilar.” Those studies lasted a median of 55 weeks (IQR: 46-78) and cost a median of $27.6 million (IQR: $18-36.7
DSCSA implementation – Down to the wire as a deadline draws near: The Drug Supply Chain Security Act (DSCSA) was enacted in 2013 as part of the Drug Quality and Security Act (DQSA), following several drug counterfeiting scandals in which falsified medical products entered the supply chain.
Research in gene therapies and genetically engineered drugs and vaccines are growing exponentially, and will only continue to become more popular. between 2020-2027. Additionally, in some cases, gene therapy treatments make permanent changes to a human’s genetic profile, which is different than typical drug treatments.
RE47,739 (‘739) by more than four years until March 5, 2027. The PTE certificate was granted under the patent restoration provisions of the Drug Price Competition and Patent Term Restoration Act of 1984. Food and Drug Administration (FDA).
Multiple drugmakers agreed to cut prices of some of their newest drugs by as much as 50% in order to receive coverage from China’s national insurance program. The companies agreed to the steep price cuts in order to have their drugs made available to the world’s second-largest pharmaceutical market. billion by 2027.
During an annual update on the program and its various workstreams, regulators and researchers discussed ongoing projects at Sentinel, its work under the current Prescription Drug User Fee authorization (PDUFA) program and next steps in using active surveillance for regulatory purposes.
Blood tends to coagulate when it comes into contact with foreign materials, and for life-support systems like ECMO to work in adults, the blood is typically thinned using drugs such as heparin. Another major issue in human babies is blood clotting.
The wide range of assertions supporting or opposing Health and Human Services’ (“HHS’”) recommendation that the Drug Enforcement Administration (“DEA”) reschedule cannabis federally from schedule I to schedule III would lead an outsider to conclude that commenters are referring to different substances. Letter to U.S.
The standards would begin with model year 2027 heavy-duty vehicles, with stringency levels through model year 2030 and beyond. This action is also supported by the President’s Executive Order 14037, titled “Strengthening American Leadership in Clean Cars and Trucks.
The standards would begin with model year 2027 heavy-duty vehicles, with stringency levels through model year 2030 and beyond. The standards will begin with model year 2027 light-duty vehicles, with stringency levels set at least through model year 2030.
March 2024 Greenhouse Gas Emissions Standards for Heavy-Duty Vehicles—Phase 3 (Final Rule Stage) 2060-AV50 On April 12, 2023, EPA announced a proposal for more stringent standards to reduce greenhouse gas emissions from HD vehicles beginning in model year (MY) 2027. 7521(a), starting with model year 2027.
This development is not just a significant milestone in drug discovery, but also a potential breakthrough for clinical practice, as it may address a critical gap in current treatments by offering effective stroke prevention with a reduced risk of bleeding. .
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.












































Let's personalize your content