This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Biosimilars have faced significant challenges in their analytical characterization. This process involves a comprehensive analysis of the biosimilar’s molecular structure, biological activity, and other quality characteristics to demonstrate similarity to the reference product. link] KBDNA. 2024, May 17). link] Lawless, L.
It allows us to make more informed decisions and identify opportunities that may have been overlooked using traditional methods.” Conducting Due Diligence Once potential targets are identified, thorough due diligence is crucial to validate the assessment and make informed in-licensing decisions.
In areas like biologics, biosimilars, and interchangeable biosimilars, where emerging technologies meet regulatory complexities, this is perhaps a wise strategy. But in the biologics and biosimilar industries, maintaining quality can be a very nuanced—if not difficult—process.
The primary vehicle for FDA to request cybersecurity information in premarket submissions has been guidance documents. Congress has given FDA the authority to require device manufacturers to provide cybersecurity information in their premarket submissions for a “cyber device.”
Lenz, Principal Medical Device Regulation Expert — FDA recently released a new eSTAR template for device pre-submissions and 513(g) Requests for Information, referred to as PreSTAR. A 513(g) Request for Information is a means of obtaining FDA’s views about the classification and regulatory requirements for a particular device.
Karst — Listing patent information in the Orange Book is a matter of judgment, but that judgment call is about to get a bit more scrutiny. The bill proposes to amend FDC Act § 505(b) and PHS Act § 351(a)(2) with respect to patent information submitted to FDA for Orange Book and Purple Book listing. Koblitz & Kurt R.
In the first category, FDA asks Congress to amend the FDCA to require drug manufacturers to disclose full information about the name and quantity of inactive ingredients in product labeling and permit FDA to disclose to generic sponsors the names and amounts of such inactive ingredients.
Shumsky — As readers of this blog know ( see, e.g. , here ), the Affordable Generics (and Biosimilars) Act has been floating around in Congress for the better part of two decades. The latest iteration of the Preserve Access to Affordable Generics and Biosimilars Act making its way through Congress is Senator Amy Klobuchar’s (D-MN) S.
Sandoz, a Novartis division, today announced progress in the late-stage clinical development program for its proposed biosimilar aflibercept. The initiation of this study marks an important milestone in the development of our biosimilar aflibercept.
Updated guidance on promotional labeling for biosimilars and interchangeables emphasizes a similar approach Today, the FDA issued a revised draft guidance on the development of promotional labeling for biosimilars, reference products, and—newly—interchangeable products. regarding its administration, preparation, storage, or safety).
This Revised Draft Guidance provides considerations for manufacturers, packers or distributors (dubbed “firms”) of prescription biological reference products, biosimilar products, and interchangeable biosimilar products presenting data and information about such products in promotional materials in a truthful and non-misleading way.
FDA’s new guidance on postapproval manufacturing changes for biosimilars focuses on current practice, new dosage forms Meeting a biosimilar user fee commitment, the FDA is expanding on its recommendations for biosimilar and interchangeable product applicants asking the FDA for post-approval manufacturing changes.
Sandoz strengthens pipeline expansion through partnership to develop and manufacture multiple biosimilars Sandoz, a global leader in off-patent (generic and biosimilar) medicines, today announced a multi-year partnership with Just – Evotec Biologics, the Seattle-based subsidiary of Evotec SE.
New FDA guidance on interchangeable biosimilar labeling heads to White House for review The FDA has submitted a draft guidance focused on the labeling of interchangeable biosimilar products to the White House for review, which would fulfill a Biosimilar User Fee Act (BsUFA III) commitment.
By Dara Katcher Levy — Yesterday, FDA published a new Draft Guidance, “ Communications from Firms to Health Care Providers Regarding Scientific Information on Unapproved Uses of Approved/Cleared Medical Products Questions and Answers ” (SIUU Guidance or Draft Guidance).
With eight marketed biosimilar medicines globally and 15+ molecules in pipeline, Sandoz is investing in future of biosimilars for patients and healthcare systems. Holzkirchen, May 3, 2021 – Sandoz, a Novartis division, today announced progress in the late-stage clinical development program for its proposed biosimilar aflibercept.
have partnered with Yangtze River Pharmaceutical Group, a leading Chinese pharmaceutical company, to form an exclusive strategic partnership for the commercialization of eight biosimilar medicines in China. is an attempt on international collaboration of biosimilar medicines. and Alvotech & CCHT Biopharmaceutical Co.,
DCI’s Outlook webinars have proven to be reliable and informative guides to crucial aspects of the ever-evolving healthcare industry. He will draw from exclusive information found in DCI's economic reports. The webinar will last 90 minutes to accommodate audience questions. Read on for full details on pricing and registration.
The update clarifies the indication by emphasizing information about the disease stages studied in the ADUHELM clinical trials. Information about the population studied has been previously communicated by Biogen and Eisai, including in the companies’ statement of June 23, 2021. Please see the full Prescribing Information.
Pharmacies and Pharmacy Benefit Managers is a definitive, nonpartisan resource that includes the most current information about pharmacy dispensing channels, third-party payers, pharmacy benefit managers (PBMs), patients’ financial contributions, government regulations, and much more. Download a free report overview for more details.
He will also draw from exclusive information found in DCI's new 2024 Economic Report on U.S. PLUS: During the webinar, Dr. Fein will give participants an opportunity to unmute themselves and ask live questions. The webinar will last 90 minutes to accommodate audience questions. Pharmacies and Pharmacy Benefit Managers.
Sandoz, a global leader in generic and biosimilar medicines, today announced the launch of a new global initiative called ‘Act4Biosimilars’ to help address health inequity and inequality worldwide. Biosimilars match their respective reference medicine in terms of quality, safety and efficacy.
Specifically, Boehringer asked FDA to interpret “strength” for biosimilars to mean “total drug content” to the exclusion of “concentration.” mL), to be biosimilar to or interchangeable with High Concentration Humira (e.g., mL), to be biosimilar to or interchangeable with High Concentration Humira (e.g.,
Click here to download a free report overview (including a summary of industry trends, the Table of Contents, and a List of Exhibits) New Drug Channels Institute Study: Biosimilars Are Delivering Higher Profits for Drug Distributors (press release) We’re offering special discounted pricing if you order before October 15, 2021! Section 4.4.
As to biosimilars, FDORA amends the statutory language regarding criteria for the demonstration of biosimilarity for a 351(k) biologic. Alternative methods will require significant research investment to demonstrate their utility for a particular context of use and inform regulatory decision-making. 42 U.S.C. § FDORA § 3209(b).
Drug Distribution Industry Expands as COVID-19 Disruption Fades and Biosimilars Boom (press release) We’re offering special discounted pricing if you order before October 21, 2022! We also update sections that were introduced in recent editions, such as our estimates of wholesalers' profits from provider-administered biosimilar drugs.
FDA unveils long-awaited Patient Medication Information proposed rule Since 2017, the FDA has been working on a proposal to create a new type of patient-focused labeling for certain outpatient drug products that would be specifically targeted for patient use. The FDA recently concluded its work on a proposed rule focused on PMI.
Bill Roth, Founding Partner, Blue Fin Group Jeff Henderson, Vice President, Head of Global Market Access, Reimbursement and Distribution, VectivBio David Weiss, Vice President, Industry Solutions, IntegriChain Rena Goins, Executive Director, Global Trade, GPO & Distribution, Regeneron Stephen Samuel, CEO, Premier Specialty Pharmacy Services Shannon (..)
Industry should take note of some of the consequential changes in the final guidance, including those related to the presentation of information about control groups, which will likely apply to many drug advertisements. FDA explained that “while some types of quantitative information are well-studied (e.g.,
BsUFA III regulatory science pilot offers progress report, fields stakeholder criticisms and questions This week, FDA and its grantees briefed stakeholders on the status of research projects funded through the Biosimilar User Fee Act (BSUFA III) Regulatory Science Pilot Program. Developing biosimilars is an extensive and expensive process.
The US Food and Drug Administration (FDA) and pharmaceutical industry groups met twice in April to discuss the reauthorization of the Biosimilar User Fee Act (BsUFA) after kicking off negotiations in March. . The two sides discussed financing, hiring and retention under BsUFA II and expectations for BsUFA III.
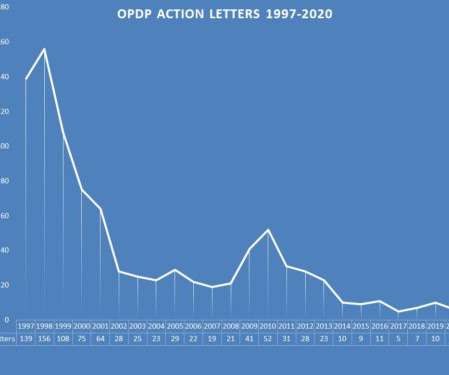
In addition, the omission or limitation of risk information – the most common subject matter of a regulatory action letter by FDA, was represented in each of the letters issued during 2020. The Violations – In recent years, there has been an uptick of activity aimed at the promotion of an unapproved drugs.
Pre-BIMO Inspection Communication Best Practices The FDA may provide a pre-announcement notice to ensure that the necessary records and personnel are available during the inspection and includes general information about what the FDA plans to review. Ask clarifying questions.
Join peers this August to discover the evolving specialty pharmacy marketplace, build multi-stakeholder partnerships, and explore frameworks to inform your 2024 distribution and patient service strategies.
AgencyIQ compiled these data using information in approval letters and review packages posted to the Drugs@FDA database. There were 21 NMEs added to the FDA’s Purple Book , which provides information about all biological products licensed by the FDA. Data on these novel approvals is published throughout the year by both CDER and CBER.
Information to include in IND, IDE, and marketing applications is also described within the draft guidance. For marketing applications, a summary of the EDDO information is recommended in the device description with reference to performance data and other supportive information.
FDA recognizes the potential of RWE to support regulatory submissions of medical devices and to inform benefit-risk analysis of such products, while assuring patients have timely access to devices. Rather than looking at how RWD/RWE provide meaningful information on safety and effectiveness, reviewers often focus on perceived gaps.
In brief, in regard to proof of identity, the seller must obtain sufficient information to identify and document the identity of the purchaser. The regulations also require detailed information about each transaction involving a tableting machine, including the date, name and address, quantity, method of transfer, etc.
Shorter exclusivity periods, driven by patent challenges and the introduction of generic or biosimilar competitors, significantly contribute to declining ROI. When a drug loses patent protection, generic or biosimilar versions can enter the market, leading to a rapid decline in sales for the original brand-name drug.
For medical devices that meet the definition of a cyber device, manufacturers are required to submit specific information in premarket submissions. Manufacturers that make modifications to an existing device will also need to submit the required information in a premarket submission. update servers, network connections, cloud, etc.)
For more information, please visit: www.innoventbio.com. The company has also entered into strategic collaborations with Eli Lilly and Company, Roche, Adimab, Incyte, MD Anderson Cancer Center, Hanmi and other international partners. View original content to download multimedia: [link]. SOURCE Innovent Biologics, Inc.
Pharmacies and Pharmacy Benefit Managers is a definitive, nonpartisan resource that includes the most current information about pharmacy dispensing channels, third-party payers, pharmacy benefit managers (PBMs), patients’ financial contributions, government regulations, and much more. This 2023 edition includes substantial new material.
While helpful background, it’s not new information. In all, the GAO Report did not provide much in the way of new information for industry. What the GAO Report does provide though is a discussion of the effect of OTC hearing aid availability on patient access to hearing loss treatment.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.
















































Let's personalize your content