This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Identifying branded drugs with a low likelihood of generic entry has become a crucial strategy for companies looking to expand their product portfolio through in-licensing. In this comprehensive guide, we’ll explore the intricacies of identifying such drugs and leveraging them for successful in-licensing opportunities.
FDA’s new guidance on postapproval manufacturing changes for biosimilars focuses on current practice, new dosage forms Meeting a biosimilar user fee commitment, the FDA is expanding on its recommendations for biosimilar and interchangeable product applicants asking the FDA for post-approval manufacturing changes.
Since 1962, the FD&C Act has authorized FDA to require that sponsors of clinical trials submit data from “preclinical tests (including tests on animals)” in order to demonstrate that their drug is safe enough to advance to testing in humans. FDORA § 3209(a)(1). FDORA § 3209(a)(2). 42 U.S.C. § 262(k)(2)(A)(i)(I). 42 U.S.C. §
Trial Also Met the Primary Endpoint in Patients With Low Levels of Eosinophils. In the subgroup of patients with baseline eosinophil counts less than 300 cells per microliter, the trial met the primary endpoint with tezepelumab demonstrating a statistically significant and clinically meaningful reduction in AAER.
This involves assessing the strength and breadth of patents, evaluating the potential for future patent challenges, and analyzing the value derived from licensing agreements and royalty streams. However, such acquisitions also entail significant risks for biotech firms.
21, 2020 /PRNewswire/ — Amgen (NASDAQ:AMGN) and AstraZeneca today announced the SOURCE trial did not meet the primary endpoint of a statistically significant reduction in the daily oral corticosteroid (OCS) dose, without loss of asthma control, with tezepelumab compared to placebo. THOUSAND OAKS, Calif. ,
Meanwhile, the FDA has also approved the Investigational New Drug (IND) application for a global Phase III trial of Toripalimab in combination with Axitinib versus Pembrolizumab for the first-line treatment of patients with advanced mucosal melanoma (Combination Clinical Trial).
Under the terms of the settlement agreement, the litigation between the parties in the United States District Court for the District of New Jersey will be ended, and Lupin will have a license to sell its generic product beginning April 2033, or earlier under certain circumstances. On March 9, 2022, the U.S. Teva Pharmaceutical Industries Ltd.
About CodeBreaK The CodeBreaK clinical trial program for Amgen’s investigational drug sotorasib is designed to treat patients with multiple KRAS G12C -mutant solid tumors and address the longstanding unmet medical need for these cancers. The Phase 2 CRC trial is expected to have a data readout in early 2021.
The length of time that it takes for us to complete clinical trials and obtain regulatory approval for product marketing has in the past varied and we expect similar variability in the future. Even when clinical trials are successful, regulatory authorities may question the sufficiency for approval of the trial endpoints we have selected.
About CodeBreaK The CodeBreaK clinical trial program for Amgen ‘s investigational drug sotorasib is designed to treat patients with an advanced solid tumor with the KRAS G12C mutation and address the longstanding unmet medical need for these cancers. The Phase 2 CRC trial is expected to have a data readout in 2021.
If the confirmatory trial shows that the drug actually provides a clinical benefit, then the FDA grants traditional approval for the drug. If the confirmatory trial does not show that the drug provides clinical benefit, FDA has regulatory procedures in place that could lead to removing the drug from the market. That did not happen.
In the third quarter, we further progressed our LEAD-EXPAND-DIVERSIFY strategy with multiple regulatory approvals, the initiation of new Phase 3 trials and the integration of the Portola team,” said Ludwig Hantson , Ph.D., Chief Executive Officer of Alexion. ” Third Quarter 2020 Financial Highlights.
The following PDUFA dates were obtained from publicly available sources. September 30, 2024 Upcoming (or overdue) legislative requirements due as of August and September Congress often asks the FDA to release or hold guidance documents, regulation, reports, meetings, hearings or pilot programs as of specific dates.
The FDA-required labeling is the drug labeling that is submitted by the sponsor and reviewed and approved by the FDA as part of the drug’s marketing application (including a New Drug Application, Abbreviated NDA, or a Biologics License Application) – and includes prescribing information (PI).
Percent on an Operational Basis, Due to Biosimilar Competition; Global Skyrizi Net Revenues Were $1.590 Billion; Global Rinvoq Net Revenues Were $731 Million. percent on an operational basis, due to biosimilar competition. Humira Net Revenues Were $16.112 Billion, an Increase of 8.4 Percent on a Reported Basis, or 12.5 Recorded a $4.7
6/27/2023 Notification FDORA, Section 3201 Within 180 days of the passage of FDORA, all biologics and biosimilars sponsors must submit a written notice to the FDA of all actively marketed products (i.e., The following PDUFA dates were obtained from publicly available sources. not discontinued) and are available for sale.
The MHRA notes that the new pathway will be parallel to its innovation pathway (the Innovative Licensing and Access Pathway , or ILAP), which integrates “early regulatory advice with health technology assessment advice.” In May 2023, the MHRA revealed which regulators will be included in the scheme: Australia, Canada, the E.U., or E.A.A.
percent on an operational basis, due to biosimilar competition. The positive CHMP opinion is based on data from the VIALE-A and M14-358 trials and represents the third positive CHMP opinion for an extension of indications for Venclyxto. Humira net revenues were $3.907 billion , an increase of 6.9 percent on a reported basis, or 12.6
Shumsky — As readers of this blog know ( see, e.g. , here ), the Affordable Generics (and Biosimilars) Act has been floating around in Congress for the better part of two decades. The latest iteration of the Preserve Access to Affordable Generics and Biosimilars Act making its way through Congress is Senator Amy Klobuchar’s (D-MN) S.
1 Ethical concerns surrounding the use of animal studies is increasing, especially considering 90 percent of drug candidates fail in clinical trials. 2 Therefore, the scientific community is researching and developing efficient ways to test compounds without the use of animals, to avoid unsuccessful outcomes in clinical trials.
All products are received in one of two forms: A New Drug Application (or NDA, for pharmaceuticals) or a Biologics License Application (BLA, for biologics). There were 21 NMEs added to the FDA’s Purple Book , which provides information about all biological products licensed by the FDA.
MacroGenics , based in Rockville, Maryland, has a target action date of December 18 for its Biologics License Application (BLA) for margetuximab in combination with chemotherapy for patients with metastatic HER2+ breast cancer. Amgen’s ABP 798, a Biosimilar to Genentech’s Rituxan. Read on for this week’s.
ENVI calls on the Commission to “revise the use of supplementary protection certificates based on technological and scientific advances to prevent generic and biosimilar medicines from becoming less competitive inside and outside the EU.”. . Denmark posts guidance on decentralizing clinical trials.
Seventeen products were orphan drugs, fourteen were generics and eight were biosimilars. Reviews of orphan medicines and biosimilars have remained stable over the past three years. There were eight biosimilars reviewed in 2023, which is comparable to 2021 (seven) and 2022 (eight), but lower than 2020, which saw 12 biosimilars reviewed.
A new real-world evidence guidance on the list is meant to address the integration of randomized controlled trials for drug and biological products into routine clinical practice. This is a topic that the agency has been keenly aware of for years, but it’s not clear from the title if it will refer to how the FDA will use A.I.
With the opinions expressed at the Advisory Committee and the data presented, the FDA will continue the review process with a decision on whether to approve the aducanumab Biologics License Application by March 7, 2021. . Biogen licensed aducanumab from Neurimmune under a collaborative development and license agreement.
Product sales increased 3% globally, driven by double digit volume growth across a number of our products including Prolia ® (denosumab), Repatha ® (evolocumab) and our biosimilar products MVASI ® (bevacizumab-awwb) and KANJINTI ® (trastuzumab-anns). AMGEVITA continued to be the most prescribed adalimumab biosimilar in Europe.
Aducanumab, an amyloid beta-targeting antibody, has been shown in clinical trials to remove amyloid beta in the brain and significantly slow clinical decline in patients with Mild Cognitive Impairment (MCI) due to Alzheimer’s disease and mild Alzheimer’s disease dementia. Since October 2017 Biogen and Eisai Co.,
The company announced donanemab received Breakthrough Therapy designation for treatment of Alzheimer’s disease and its intention to submit a biologics license application (BLA) for donanemab under the accelerated approval pathway later this year based on data from TRAILBLAZER-ALZ. Revenue outside the U.S. decreased 28 percent to $95.6
Biogen licensed aducanumab from Neurimmune under a collaborative development and license agreement. Results in early stage clinical trials may not be indicative of full results or results from later stage or larger scale clinical trials and do not ensure regulatory approval. Since October 2017 Biogen and Eisai Co.,
and Annex 1 Conference Joel Welch December 18 RAPS RAPS Webcast: FDA Forecast: What’s Next for the FDA in 2024 AgencyIQ Speakers December 21 HL7 REMS Public Call PDUFA Dates expected in November and December PDUFA dates represent the expected date of a regulatory decision by the FDA on a New Drug Application or Biologics License Application.
Novartis has been granted an option to in-license global rights of MP0420 and MP0423 – multi-targeted direct-acting antiviral therapeutic candidates demonstrating potential efficacy against COVID-19.
Upon option exercise, Novartis would be responsible for all further development and commercialization activities.
Nasdaq: BIIB) and MedRhythms have entered into a license agreement to develop and commercialize MR-004, an investigational prescription digital therapeutic for the potential treatment of gait deficits in multiple sclerosis (MS). Biogen Inc.
September 2023 Final Rule Stage Biologics License Applications and Master Files The Biologics Price Competition and Innovation Act of 2009 (BPCI Act) deemed any biological product approved in a new drug application (NDA) to be a biologics license application (BLA) on March 23, 2020. That would leave 19% of staff affected by furloughs.
Title Type Comments Close Classification Categories for Certain Supplements Under BsUFA III Guidance October 10 Formal Meetings Between the Food and Drug Administration and Sponsors or Applicants of Biosimilar User Fee Act Products Guidance October 10 QTc Information in Human Prescription Drug and Biological Product Labeling Guidance October 10 Postmarketing (..)
We continue to work in parallel on our two COVID-19 vaccine candidates, with clinical trials starting in the coming weeks. In Europe, where the product is commercially available in several countries and has a temporary license to be sold in France, sales were €12 million. Full-year 2020 franchise sales were down 4.8%
We continue to work in parallel on our two COVID-19 vaccine candidates, with clinical trials starting in the coming weeks. In Europe, where the product is commercially available in several countries and has a temporary license to be sold in France, sales were €12 million. Full-year 2020 franchise sales were down 4.8%
reflecting increased competition partially offset by demand growth partly related to clinical trial supply and price upside in Europe. net price, increasing usage of Toujeo ® , biosimilar glargine competition and lower sales in Europe (patient stockpilling in the first quarter of 2020). Aubagio ® sales decreased 1.1% In the U.S.,
12/29/2023 FDORA, Section 3602 Clinical Trials Modernization : FDA is directed to require the submission of a “diversity action plan” for all Phase 3 clinical trials of new drugs. ” These include the use of expansion cohorts, concurrent trial conduct, and other designs. .”
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.








































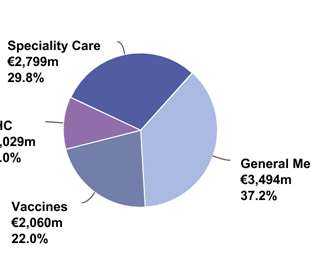
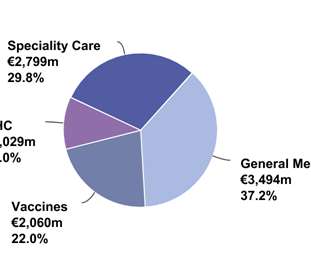
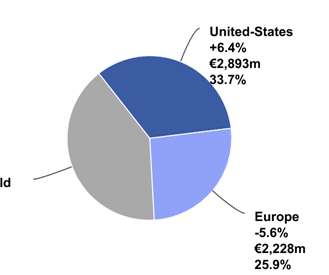






Let's personalize your content