This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The rest of this blog will focus on the Clinical Trials Grants Program. The FDA’s Orphan Products Clinical Trials Grants Program is open to academic institutions, industry sponsors, non-profit organizations, and public or private entities, both within and outside the U.S. Relative to other areas of medicine (e.g.,
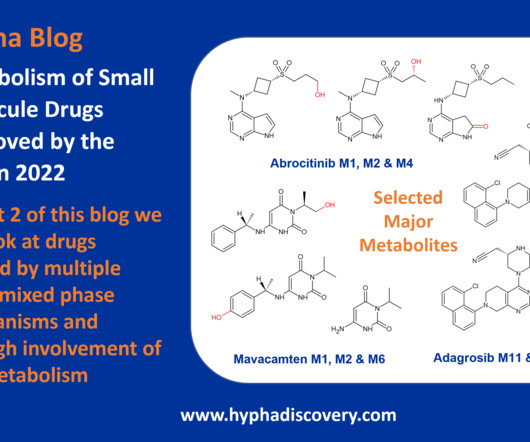
Metabolism of 2022 FDAapproved small molecule drugs part 2 Mixing it Up By Julia Shanu-Wilson In Part 1 of this topic we looked at metabolism of the small molecule drugs approved by the FDA in 2022 that were mediated by CYP3A4. Dermavant’s tapinarof is one such friend. 8 This is not the only point of interest.
Livornese — I saw the sign…and the answer is no—FDA-approved labeling apparently is not enough under state failure-to-warn laws, according to certain courts. The GAO Report further explained that the agency did not have the resources to regulate the estimated 100,000 OTC drugs marketed through the monograph process.
Valentine — On November 22, 2022, FDAapproved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
Effective and consistent use and application of Data Standards can reduce costs of Pharmaceutical Drug and Biologic Products and Process Development, Drug Development Services, 505(b) NDA, IND Consulting , NDA Consulting, BLA Consulting , and effective FDA Pre-Submission collectively resulting in FDAApproval. Spanogle, Ph.D.
Effective use of Real World Data (RWD) and Real World Evidence (RWE) can reduce costs of Pharmaceutical Drug and Biologic Products and Process Development, Drug Development Services , expedite a FDA Pre-Submission Review, and lead to FDAApproval. Author Information William E. Spanogle, Ph.D.
The American Conference Institute (“ACI”) will be hosting the go-to forum for critical updates on OTC regulation and enforcement, monograph reform, ACNU and advertising essentials… and FDA Law Blog readers can get a discount. FDA Law Blog is a conference media partner for this event.
FTC, deep in its foray into the Orange Book, filed an Amicus Brief in the case arguing that the patents do not claim any FDA-approved drug. While the relevant regulation defines “drug product” as the “finished dosage form,” which theoretically includes the delivery device, the Court again falls back on the wording in 21 C.F.R.
ENTRESTO was approved by FDA in July 2015 “to reduce the risk of cardiovascular death and hospitalization for heart failure in patients with chronic heart failure (NYHA Class II-IV) and reduced ejection fraction.”
The Act is intended to address national security concerns by prohibiting certain conduct by regulated industry. Effect on FDAapprovals— In cases where a biological product or drug needs to change aspects of its manufacturing processes to avoid using a covered equipment or service, will it need to file supplements with FDA for the CMC update?
Karst — It’s been a while since we last blogged on Patent Term Extension (“PTE”) issues of interest. 156, a patent may be extended only once (even if it would be eligible for extension on more than one occasion because it applies to several FDA-approved products), and only one patent may be extended for each regulatory review period.
Governors, state cannabis regulators, law enforcement groups and local governments weighed in, as did marijuana advocates and opponents, marijuana industry associations, Members of Congress, federal law enforcement groups, healthcare and human rights groups, unions and trade associations, and private individuals.
Dr. Won will share his expertise and insights on the intricacies of the United States Food and Drug Administration (FDA) regulations pertaining to medical devices, with a special focus on class II and class III hearing devices. Philip Won , of Hyman, Phelps & McNamara, P.C.
Clinical studies are often conducted to support a Premarket Approval (PMA) application, though some 510(k) submissions may require clinical data. In this blog post, we discuss key considerations for assessing the need for an IDE and complying with the reporting requirements under the IDE program. 2 Existence of a predicate device.
In 2016, the Food and Drug Administration (FDA) approved Spinraza (nusinersen). While the FDA’sapproval of nusinersen may not seem extraordinary, it was. Nusinersen’s approval marked the first time nonclinical data supported conducting initial clinical trials involving children. Why This Guidance Now?
Palmer — A new lawsuit against FDA is the latest happening in the veterinary drugs space and, by extension, FDA’s Center for Veterinary Medicine (CVM). We blogged about CVM last week and explained the increasing attention to animal health products due to the expansion of the animal and pet product market. Koblitz & Karla L.
Karst — If you’ve been following this blog since the early days, then you know we fervently followed the more-than-decade-long soap opera that was The Medicines Company’s efforts to obtain a Patent Term Extension (“PTE”) from the U.S. Usually at this point in a post we would identify the date of approval of the relevant NDA.
In this blog, we examine how mRNA can impact cancer treatment, the unique challenges associated with working with mRNA, and strategies for researchers proposing mRNA-based cancer trials. Many monoclonal antibodies are used to treat cancer by targeting specific cell structures regulating cell activity, causing the death of tumor cells.
FDA has not approved an NDA for a drug containing botanical marijuana but notes that two drug products containing delta-9-tetrahydrocannabinol (“delta-9-THC”) (as dronabinol), the primary compound in marijuana have received FDAapproval: Marinol and Syndros. chemotherapy-induced), and pain.
It merely says that “[t]he Hatch-Waxman Act and FDAregulations set forth the criteria for listing patents in the Orange Book” and that “Brand manufacturers are responsible for ensuring their patents are properly listed.” But it is not always clear which types of patents are eligible for listing in the Orange Book.
To date, the only FDAapproved immune checkpoint inhibitors targeting the PD-1/PD-L1 pathway are monoclonal antibodies (mAbs). The post Researchers Identify Novel Immunotherapeutic Compound Targeting Key Cancer Pathway first appeared on PerkinElmer Blog.
As with other FDA-regulated products, such as human drugs and medical devices, the “regulatory review period” is composed of a “testing phase” and a “review phase.” The “review phase” is the period between the initial submission and approval of the NADA. FDA’s PTE regulations at 21 C.F.R.
By Riëtte van Laack — For those readers unfamiliar with the regulation of animal food ingredients in the United States, below is a brief background. In the United States, animal food (feed) regulations are enforced by state and federal regulatory officials. AAFCO develops model regulations, that serve as model for the states.
The ramifications of the Federal Claims case and some of the other lawsuits brought by Vanda could be significant to regulated industry and to the food and drug bar. FOOD AND DRUG ADMINISTRATION et al Challenge to FDAapproval of generic Hetlioz (tasimelteon) Pending 1:2023cv00629 (COFC) VANDA PHARMACEUTICALS, INC.
Molecular Weight: 631.700 FDAAPPROVED, To treat moderately to severely active ulcerative colitis in adults, 10/12/2023 Velsipity Etrasimod , sold under the brand name Velsipity , is a medication that is used for the treatment of ulcerative colitis (UC). [1] “FDAApproves New Drug for Ulcerative Colitis” Medscape.
Since 1982, when the Federal Drug Administration (FDA) approved the first recombinant biotechnology drug, insulin, for human use, biotechnology, particularly biologic entities and biological processes, has provided key inputs for the development of pharmaceutical goods.
Gaulkin — In May 2023, we posted about a CMS proposed regulation that sought to make a wide variety of changes to the Medicaid Drug Rebate Program (MDRP), including a new “price verification survey,” and a controversial proposal to require “stacking” of discounts to different customers when determining best price.
The treatment had been granted breakthrough therapy designation, but FDAapproval would ultimately rest on the sponsor’s ability to demonstrate clinically meaningful improvement. Key Takeaway RWD and RWE play an important role in rare diseases, where capturing relevant data to support drug development is challenging.
This feedback prompted the FDA to release a second paper in 2021 titled “ Artificial Intelligence/Machine Learning (AI.ML)-Based Software as a Medical Device (SaMD) Action Plan ,” which outlined the agency’s five-pillar approach to regulating AI/ML.
1 Factor B is a positive regulator of the alternative complement pathway, where it activates C3 convertase and subsequently C5 convertase. 2 This is of particular importance to PNH, where one of the disease hallmarks is the mutation of the PIGA gene. 1 H NMR (400 MHz, methanol-d4) δ 10.73 (s, 1H), 8.23 (d, J = 8.2 Hz, 2H), 7.74 (d, J = 8.3
Approximately six weeks after FDAapproved Lumryz and issued its clinical superiority decision , Jazz filed a Complaint against FDA in the District Court of D.C. Jazz also argues that, notwithstanding the language that instructs FDA to promulgate regulations implementing the clinical superiority provisions at 21 U.S.C.
FDA provided detailed support, going through almost every instance that Boehringer cites as evidence that FDA interpreted “strength” differently in 2009, to show that FDA has consistently applied the same definition of “strength” since the enactment of the Hatch-Waxman Act. do not help Boehringer’s case.
In fact, federal law and regulation appear to require AbbVie to list these patents.” No matter how Congress and FTC frame it, it is FDA that’s to blame for the listing confusion when it has had more than 20 years to respond to questions about whether these types of patents should have been listable in the first place.
Because a “drug product” is defined by regulation as a “Finished dosage form. that contains a drug substance ,” FTC explains that “only those [patents] that claim the finished dosage form containing the drug substance of the relevant NDA” can be listed.
Years of hard work, supported by the National Institutes of Health and the Cystic Fibrosis Foundation, painstakingly worked out the normal function of the protein that is altered in CF, called the cystic fibrosis transmembrane regulator (CFTR). Credit: Zhang & Chen, 2016, Cell 167, 1586–1597. About 30,000 Americans have CF.
1067, the “ Ensuring Timely Access to Generics Act of 2023 ,” and it would fundamentally transform the playing field for NDA, ANDA, BLA, and aBLA applicants seeking to preserve their rights in the wake of an adverse FDAapproval decision. That bill is S. The fact that S. 1067 at the earliest possible opportunity.
Food and Drug Administration – labeled an “Official Action Indicated” classification – is generally devastating for the facility, not least because it can stall FDAapproval of applications to market drugs manufactured at the facility. Farquhar — A drug manufacturer’s bad post-inspection grade from the U.S.
This statutory provision requires generic drugs to have the same labeling as that approved for the listed drug “except for changes required because of differences approved under a [suitability petition, allowing for certain differences] or because the new drug and the listed drug are produced or distributed by different manufacturers.”
156, as added by the 1984 Hatch-Waxman Amendments, for certain FDA-regulated products. The use of a surrogate endpoint can considerably shorten the time required prior to receiving FDAapproval. Karst — Earlier this week, we posted Part 1 of our three-part series on U.S.
Read Small-molecule aptamer for regulating RNA functions in mammalian cells and animals. Read Eisai and Biogen — companies behind the controversial, FDA-approved drug for Alzheimer’s, called Aduhelm — have published three additional papers with “detailed analyses” from their phase 2 trials.
OASH concluded “there is widespread current experience with medical use of marijuana in the United States” by licensed healthcare providers “operating in accordance with implemented state-authorized programs, where such medical use is recognized by entities that regulate the practice of medicine under these state jurisdictions.”
156 for certain FDA-regulated products, we know what you were thinking. 156(d) (and the PTO’s PTE regulations at 21 C.F.R. That regulation is pretty clear. On April 29, 2022, FDAapproved Tap Pharmaceuticals, AG’s (“Tap’s”) NDA 215809 for EMERZA (levothyroxine sodium) Oral Solution. 2d 1220, 1223 (Fed.
FDAapproves 100th monoclonal antibody product ( Nature Reviews Drug Discovery ).
Inside Vida Health’s $110M Series D & Big Push into Digital Mental Health ( The Health Care Blog ).
AI, digital health feature in latest batch of FDA breakthrough device designations ( MedTech Dive ).
While denying a Citizen Petition from Novartis asking FDA to refuse to approve any ANDA that omits the new dosing regimen or changes the indication, FDAapproved MSN’s ANDA on July 24, 2024.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.


















































Let's personalize your content