This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Growing acceptance of biomarkers by regulatory bodies Regulatory guidelines are just as crucial in early development as in later stages, though they allow for more flexibility in endpoints and investigational strategies. Biomarkers can play a crucial role throughout clinicaldevelopment, especially in early phases.
Food and Drug Administration (FDA) plays a pivotal role in fostering the development of treatments for rare diseases through its Orphan Products Grants Program. Each year, FDA selects a limited number of clinical trials to fund to help sponsors pursue development of medical products for rare diseases and advance their field.
There are over 7,000 rare diseases affecting more than 30 million people in the United States, and despite the FDA's approval of over 600 treatments for rare diseases since signing the Orphan Drug Act into law in 1983, most rare diseases still do not have a treatment.
Food and Drug Administration (FDA) that is expected to continue to rise, it’s critical to find methods that materially help improve disease outcomes. Data, technology and AI are being used in innovative ways to optimize study design and trial protocol development and predictive analytics.
Patients are the backbone of clinical trials, playing an essential role in the drug development process. However, patients also play a vital role in engaging directly with the FDA. It also means sponsors are less likely to waste time and resources developing programs that don’t work for patients.”
Researchers must characterize the anti-drug-antibody (ADA) response in preclinical and clinical studies and report any ADA-positive samples as a risk-based approach. Regulatory Considerations for Oligonucleotide Drug Development and Safety In 2024, the U.S.
AlgoTherapeutix recently raised a 12M€ Series A that will fund the Phase 1 and 2 clinicaldevelopment of ATX01. In parallel, AlgoTx firmed-up ATX01’s development pathway via a pre-IND consultation with the FDA and obtained an Orphan Drug Designation from the FDA to explore ATX01’s activity in erythromelalgia.
Nitrosamines are hardly ever good, Now FDA has issued policy. All of these deviations discussed in FDA inspectional observations and Warning Letters have caused serious issues for manufacturers of APIs or finished drug products. An earlier FDA guidance, revised in 2021. An earlier FDA guidance, revised in 2021.
Preliminary blinded data on NVX-CoV2373 in older adults needed to proceed to Phase 3 has previously been positively reviewed by the Food and Drug Administration (FDA). Additional clinical data from the Phase 2 trial conducted in the U.S. NVX-CoV2373 (SARS-CoV-2 vaccine) FDA Approval History. Source: Novavax, Inc. .
Food and Drug Administration (FDA) for the treatment of adult patients with deleterious or suspected deleterious BRCA -mutated ( BRCA m) metastatic castration-resistant prostate cancer (mCRPC). Patients should be selected for therapy based on an FDA-approved companion diagnostic for LYNPARZA. In the U.S., For the U.S.
The US Food and Drug Administration (FDA) has accepted Roche’s application for a new self-administration option for Xolair (omalizumab) across all approved US indications. Its use in these settings is supported by a rigorous clinicaldevelopment programme, including eight Phase III studies.
Source link.
Clissold — The trio of CDER, CBER, and CDRH released a new draft guidance titled “ Use of Data Monitoring Committees in Clinical Trials ” that revises the 2006 guidance “Establishment and Operation of Clinical Trial Data Monitoring Committees” and, when final, will replace the 2006 guidance.
Read the Guide FDA Launches Pilot Program to Help Further Accelerate Development of Rare Disease Therapies One frustrating aspect of traditional drug development, especially for rare disease communities, is the tempo of regulatory decisions on potential drugs.
and Shionogi Limited as shareholders, today announced the positive findings of a pooled analysis of COVID-19-related impacts across the investigational long-acting cabotegravir and rilpivirine clinicaldevelopment programme. Related Articles: Cabenuva (cabotegravir and rilpivirine) FDA Approval History. Source: GSK .
FDA Under Emergency Use Authorization.
FDA that Emergency Use Authorization (EUA) submission is acceptable for all three COVID diagnostic tests. SQI intends to submit RALI-dx for EUA to FDA in late Q4 2020. SQI intends to submit RALI- fast for EUA to FDA in late Q1 2021.
TORONTO , Oct.
Regulatory bodies such as the FDA oversee clinical trials to ensure that studies’ design, conduction, analysis, and reporting are per established guidelines and laws. Rigorous procedures to ensure that drugs are effective and safe.
Food and Drug Administration (FDA) for the development of LX9211 in diabetic peripheral neuropathic pain. The FDA’s Fast Track designation of LX9211 reflects the serious unmet medical need of people suffering from diabetic peripheral neuropathic pain,” said Praveen Tyle, Ph.D., THE WOODLANDS, Texas, Dec. Safe Harbor Statement.
Data is the cornerstone of any clinical trial, driving the decision-making process of drug development, and is a fundamental requirement for approval of new therapies by regulatory agencies like the FDA or EMA.
Even though AI-designed drugs arent yet a household term for FDA-approved, commercially available therapies, they are a reality in clinicaldevelopment pipelines. Even though AI-designed drugs arent yet a household term for FDA-approved, commercially available therapies, they are a reality in clinicaldevelopment pipelines.
The Washington Nationals won the World Series, Presidential administrations have come and gone, and FDA has added new meeting types and formats to its menu. And so, FDA has issued a new draft guidance to bring everyone up to speed on formal meetings under PDUFA. Tobolowsky — Much has changed since the long-gone days of 2017.
Speakers discussed investigator, regulatory (FDA), industry, and patient perspectives during the special symposium “Challenging the Status Quo of Early Phase Clinical Trial Design: Project Optimus.” To address this challenge, the FDA Oncology Center of Excellence initiated Project Optimus.
Food and Drug Administration (FDA) granted Breakthrough Therapy Designation (BTD) to TAK-994, 1 its Phase 2 investigational oral orexin agonist, which is designed to selectively target orexin 2 receptors. – If Approved, Investigational TAK-994 May Provide a Future Treatment Option Targeting the Orexin Deficiency Underlying NT1.
The FDA has approved a request from American Gene Technologies to begin a clinical study into its HIV gene therapy. In a paper published in Molecular Therapy: Methods & ClinicalDevelopment , the team discussed why they choose this type of gene therapy to treat HIV. Air Force photo by Kemberly Groue.
Food and Drug Administration (FDA) has converted this indication from an accelerated to a full (regular) approval. 10), as determined by an FDA-approved test, or in patients who were not eligible for any platinum-containing chemotherapy regardless of PD-L1 status.
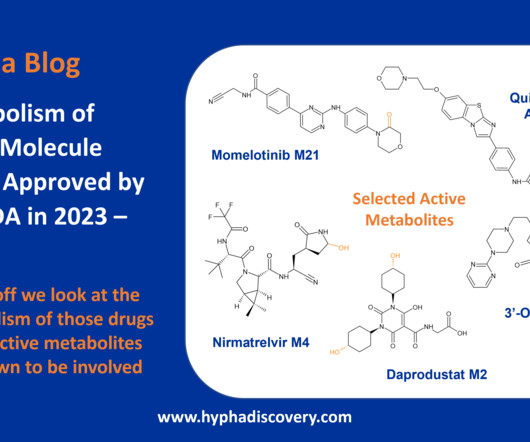
Metabolism of 2023 FDA Approved Small Molecules – PART 1 By Julia Shanu-Wilson 2023 was a fruitful year for drug approvals by the FDA, with a crop of 34 small molecules out of a total of 55 new drugs [1]. are major metabolites according the FDA Metabolites in Safety Testing guidelines). 2] Iversen et al.,
Food and Drug Administration (“FDA”) has approved the Investigation New Drug (IND) application for TG-1000, a novel treatment for influenza A and B. Taigexyn ® is already on the market in the mainland China and Taiwan , TG-3000 has completed Phase 2 clinical studies, and Furaprevir is currently in Phase 3 clinicaldevelopment.
Food and Drug Administration (FDA) has lifted the clinical hold placed on the company’s Investigational New Drug Application (IND) to evaluate injectable lenacapavir for HIV treatment and HIV pre-exposure prophylaxis (PrEP). (Nasdaq: GILD) today announced the U.S.
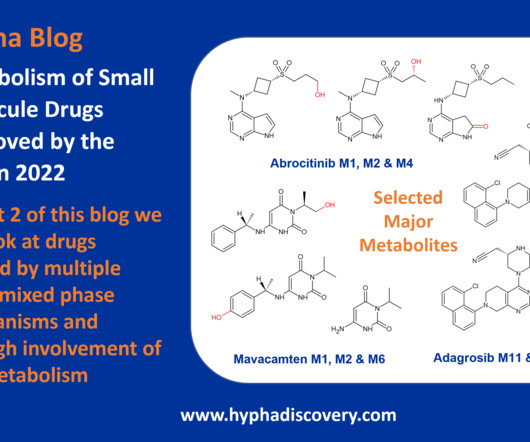
Metabolism of 2022 FDA approved small molecule drugs part 2 Mixing it Up By Julia Shanu-Wilson In Part 1 of this topic we looked at metabolism of the small molecule drugs approved by the FDA in 2022 that were mediated by CYP3A4. 16 This concludes our exploration of metabolism of last year’s FDA approved small molecule drugs.
Metabolism of 2022 FDA approved small molecule drugs – Part 1 Does CYP3A4 still rule? By Julia Shanu-Wilson It won’t come as much surprise to learn that of the 17 small molecules* approved by the FDA in 2022, CYP3A4 was the major player in drug metabolism. References Iversen et al., Front Pharmacol.,
NASDAQ: HARP), a clinical-stage immunotherapy company developing a novel class of T cell engagers, today announced that the U.S. Food and Drug Administration (FDA) has granted Orphan Drug Designation for HPN217 for the treatment of multiple myeloma. SOUTH SAN FRANCISCO, Calif., About the Phase 1/2 Trial for HPN217.
(Nasdaq: NVAX), a late-stage biotechnology company developing next-generation vaccines for serious infectious diseases, today announced that the U.S. Food and Drug Administration (FDA) has granted Fast Track Designation for NVX-CoV2373, the Company’s COVID-19 vaccine candidate. NVX-CoV2373 (SARS-CoV-2 vaccine) FDA Approval History.
Jerry has over 30 years of experience in the biopharmaceutical industry and has been involved in the discovery, clinicaldevelopment, and global commercialisation of more than a dozen FDA-approved drugs with multiple successful exits. About the author Jerry McLaughlin CEO, Life Biosciences, Inc.
Recent correspondence between the Company and the FDA resulted in modifications to the previously disclosed trial design, including designating overall survival (OS) as the primary endpoint of the study. Based on this, the trial has the potential to provide data to the FDA that may allow an expedited pathway for development.
7, 2021 /PRNewswire/ — UNION therapeutics A/S ( UNION ) today announces that the US Food and Drug Administration (FDA) has approved an Investigational New Drug program (IND) for oral orismilast; a next generation PDE4-inhibitor for the treatment of plaque psoriasis in adults.
HELLERUP, Denmark , Jan. Sources 1: Li et al.
Data monitoring committees (DMCs) review ongoing clinical trial data to make recommendations regarding trial conduct based on risk-benefit. DMCs are an essential component to ensuring the integrity and safety of clinical trials. New draft guidance published by the U.S. The draft is open to public comment until April 15, 2024.
Legislation on pediatric studies has existed for more than 20 years in the US, yet additional guidance from the FDA has been relatively scarce. How might this affect pediatric drug development?
Food and Drug Administration (FDA). Teva will continue to lead the clinicaldevelopment and regulatory process and be responsible for commercialization of this candidate treatment, with MedinCell eligible for development milestones, royalties on net sales and future commercial milestones.
For instance, Inflectra , a biosimilar of Remicade, underwent a comprehensive clinicaldevelopment program, including a Phase III trial demonstrating comparable efficacy and safety to the reference product in patients with rheumatoid arthritis. The European Medicines Agency (EMA) and the U.S.
During clinicaldevelopment, new chemical entities (NCEs) require an absorption, metabolism, and excretion (AME) study. The post Why Proactive AME Studies are Critical to Accelerating Your Approval Journey appeared first on Worldwide Clinical Trials.
14, 2021 /PRNewswire/ — MindMed (NEO: MMED), (OTCQB: MMEDF), (DE: MMQ), a leading psychedelic medicine biotech company today announced the addition of Robert Barrow , an accomplished pharmaceutical executive, as Chief Development Officer. We are excited to attract such top tier talent from the psychedelic drug development community.
Food and Drug Administration (FDA) made public a potentially game-changing proposal concerning the regulatory framework for laboratory-developed tests (LDTs). Understanding the nuances and implications of these changes is paramount for specialists in regulatory affairs and the clinicaldevelopment arena. billion and $86.01
Quizartinib NDA Review for Patients with Newly Diagnosed FLT3- ITD Positive AML Extended by FDA Daiichi Sankyo (TSE: 4568) today announced that the U.S. No additional efficacy or safety data has been requested. “We
His presentation covered various aspects of oncology clinical programs, focusing on study design trends, with reference to both the recently implemented FDA Project Optimus guidance and studies we have seen from sponsors.
(Nasdaq: PASG), a genetic medicines company focused on developing transformative therapies for rare, monogenic central nervous system (CNS) disorders, today announced that the U.S. We are excited to investigate the potential of PBFT02 as a treatment for FTD-GRN as we initiate our clinicaldevelopment program in the coming months.”.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.













































Let's personalize your content