This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The trial, which will explore the pharmacokinetics and safety of ATX01 in healthy volunteers, is due to start in January 2021. AlgoTherapeutix recently raised a 12M€ Series A that will fund the Phase 1 and 2 clinicaldevelopment of ATX01. More information at www.algotx.com. View source version on businesswire.com: [link].
Regulatory bodies such as the FDA oversee clinical trials to ensure that studies’ design, conduction, analysis, and reporting are per established guidelines and laws. Rigorous procedures to ensure that drugs are effective and safe.
Food and Drug Administration (FDA) ’s focus appears to be on Emergency Use Authorizations (EUAs) for the Pfizer-BioNTech and Moderna COVID-19 vaccines, as the year wraps up there are still some PDUFA dates on the agency’s calendar. Zai Lab is leading clinicaldevelopment in its territory. “We Although much of the U.S.
His presentation covered various aspects of oncology clinical programs, focusing on study design trends, with reference to both the recently implemented FDA Project Optimus guidance and studies we have seen from sponsors.
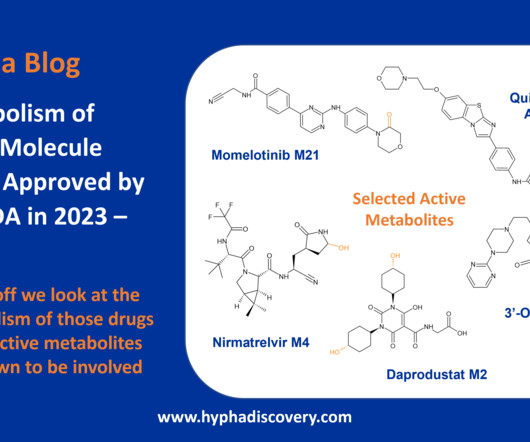
Metabolism of 2023 FDA Approved Small Molecules – PART 1 By Julia Shanu-Wilson 2023 was a fruitful year for drug approvals by the FDA, with a crop of 34 small molecules out of a total of 55 new drugs [1]. are major metabolites according the FDA Metabolites in Safety Testing guidelines). 2] Iversen et al.,
Food and Drug Administration (FDA) in terms of PDUFA dates. Here’s a look at some of the upcoming target action dates on the FDA’s calendar. . The resubmission came after a meeting with the FDA in February 2020 and addresses issues raised by the agency’s Complete Response Letter (CRL) in November 2019. “We
UBX1325 is developed from BM-962, a Bcl-xL inhibiting compound licensed to UNITY by Ascentage Pharma for the treatment of age-related diseases. This progress in clinicaldevelopment qualifies Ascentage Pharma for a milestone payment according to the terms of the licensing agreement. SUZHOU, China and ROCKVILLE, Md. ,
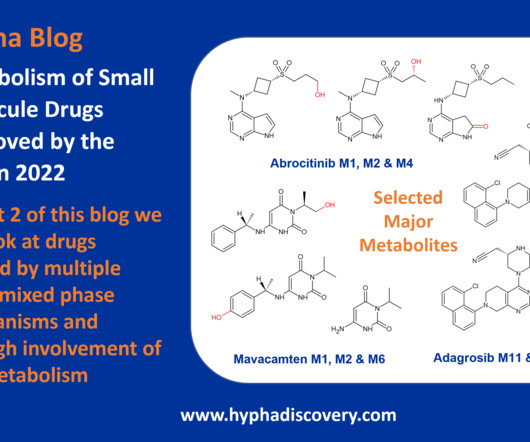
Metabolism of 2022 FDA approved small molecule drugs part 2 Mixing it Up By Julia Shanu-Wilson In Part 1 of this topic we looked at metabolism of the small molecule drugs approved by the FDA in 2022 that were mediated by CYP3A4. 16 This concludes our exploration of metabolism of last year’s FDA approved small molecule drugs.
In this blog, we explain the role of clinical pharmacology in drug development and demonstrate how the right strategy can accelerate development under the US Food and Drug Administration (FDA) 505(b)(1) and 505(b)(2) New Drug Application (NDA) pathways. Author: Jayesh Patel , Principal Scientist, Pharmacokinetics
The FDA EUA submission is based on an interim analysis of efficacy and safety data from the Phase 3 COMET-ICE (COVID-19 Monoclonal antibody Efficacy Trial – Intent to Care Early) trial, which evaluated VIR-7831 as monotherapy for the early treatment of COVID-19 in adults at high risk of hospitalisation.
BY AMANDA CONTI, ALEXANDER GAFFNEY, MS, RAC SEP 18, 2023 9:24 PM CDT Background: FDA’s standards for evidence When seeking approval from the FDA, companies are required to demonstrate that their product is safe and effective when used as intended. In one landmark case, Warner-Lambert Co.
Sisunatovir significantly reduced viral load in a phase 2 RSV human challenge study in healthy adults and is currently in phase 2 clinicaldevelopment in infants. The development program for sisunatovir is expected to continue in both adult and pediatric populations. Food and Drug Administration (FDA).
The pharmaceutical industry is under huge pressure to address the high attrition rates in drug development. With around 90% of candidates failing during clinicaldevelopment, 1 the process is not only long and risky, but also expensive for those involved. References Hingorani, A.D., Sci Rep 9, 18911 (2019).
Aside from the advent of complex antibody-based drugs, the industry is facing some additional changes which are shaping drug development. Recently, the FDA announced that new medicines need not be tested in animals to receive regulatory approval. FDA no longer needs to require animal tests before human drug trials [Internet].
Food and Drug Administration (FDA) clearance of an Investigational New Drug (IND) application for SLV213 for the treatment of COVID-19 and has dosed the first subjects in a Phase 1 clinical study. Preclinical data show SLV213 is a potent inhibitor of SARS-CoV-2 infection. announced today that the company has received U.S.
There is a continuum of evidence for a given target – at one end are novel targets with some evidence of importance in disease, and at the other end are “de-risked” targets where the biology is precedented with an approved product or late-stage clinical asset(s).
Food and Drug Administration (FDA) as a nasal spray called esketamine, for treatment-resistant depression. Since the FDA approved the first oligonucleotide drug for clinical use in 1998, to treat cytomegalovirus retinitis (CMV) in immunocompromised patients, the therapeutic landscape has undergone significant change.
Food and Drug Administration (“FDA”) for the treatment of agitation associated with delirium. In addition, this indication offers synergy with the commercial infrastructure being developed to support our first New Drug Application.”. The Company plans to initiate a Phase 2 trial within the next several months. “We
FDA finalizes new guidance on nonclinical assessment of potential immunotoxicity Late last week the FDA released a final guidance document containing recommendations on the nonclinical assessment of potential immunotoxicity for drugs and certain biologics.
The FDA and oncology: 2023 year in review As we round the corner into the last few weeks of 2023, AgencyIQ has taken a look back at a very busy year for the FDA’s oncology staff, and for sponsors. Since its inception, OCE has initiated an extensive list of initiatives and has taken the lead on over forty guidances published by FDA.
(Nasdaq: INZY), a clinical-stage biopharmaceutical company developing novel therapeutics for the treatment of rare diseases of abnormal mineralization impacting the vasculature, soft tissue and skeleton, today announced that the U.S.
About the study NCT03832114 is a Phase II, open- marker, two cohort,non-randomized study assessing the efficacity, safety and pharmacokinetics of iptacopan in cases with C3 glomerulopathy (C3G) ( cohort A) and cases who have experienced order transplantation and have posterior C3G rush in the transplanted organ ( cohort B).
” The Phase III clinicaldevelopment program consists of three studies, the BRIDGE study, the BALANCE study and the BRIGHT study. Food and Drug Administration (FDA) for pegunigalsidase alfa for the proposed treatment of adult patients with Fabry disease via the FDA’s Accelerated Approval pathway.
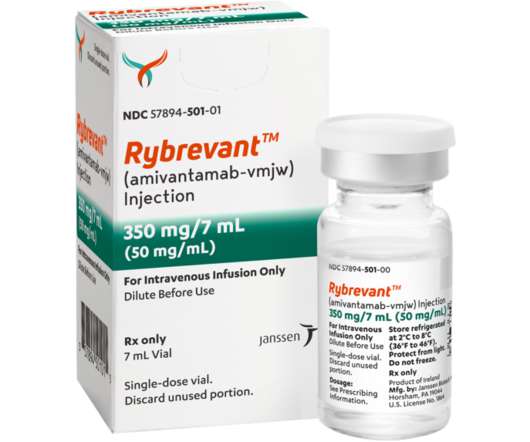
Food and Drug Administration (FDA) approval for patients with locally advanced or metastatic NSCLC with epidermal growth factor receptor (EGFR) exon 20 insertion mutations, based on data showing an ORR of 40 percent (95 percent CI, 29 – 51) and median duration of response of 11.1 months (95 percent CI, 6.9 – NE). [7]. About RYBREVANT TM.
of new cancer drugs tested in Phase I were likely to receive Food and Drug Administration (FDA) approval. Additionally, in 2021, a literature review estimated the average capitalized research and development (R&D) costs per new cancer medicine at between $944 million and $4.54
We ’re agitated to partake data from our robust clinicaldevelopment programs at AASLD’s The Liver Meeting ®, including the rearmost data demonstrating the positive impacts of bulevirtide for people living with HDV.”. Food and Drug Administration (FDA). Bulevirtide is an investigational agent in theU.S.
In late October, SetPoint Medical received Breakthrough Device Designation from the FDA for the use of its novel bioelectronic device by patients with rheumatoid arthritis (RA) who have incomplete response to, or are intolerant to multiple biologic drugs. The secondary outcome involves pharmacokinetic endpoints. Bioelectronic Platform.
18, 2021 /PRNewswire/ — Genkyotex SA , a subsidiary of Calliditas Therapeutics AB (publ) (“Calliditas”) (Nasdaq OMX – CALTX; NASDAQ – CALT), today announced positive Phase 1 data demonstrating a favorable safety and pharmacokinetic profile of high-dose setanaxib, Genkyotex’s lead asset. STOCKHOLM , Jan.
“We expect 2021 will mark a number of key clinical and commercial milestones. By the end of the year, we expect to have five independent TransCon product candidates in clinicaldevelopment leveraging TransCon technologies through our algorithm for product innovation. Pipeline Updates.
•.
?.
FDA’s Diversity Action Plan: Questions, answers, and what we (don’t) know so far Last month, the FDA published a new draft guidance document on diversity action plans. The law also directed the FDA to “update or issue guidance” on DAPs. Read AgencyIQ’s extensive analysis of the draft guidance here.]
FDA’s new guidance on postapproval manufacturing changes for biosimilars focuses on current practice, new dosage forms Meeting a biosimilar user fee commitment, the FDA is expanding on its recommendations for biosimilar and interchangeable product applicants asking the FDA for post-approval manufacturing changes.
First-in-class LNP023 – which potently and selectively targets factor B, part of the immune system’s alternative complement pathway 3,4,5 – is also in development for several renal conditions with complement system involvement . The FDA and EMA have granted orphan drug designations to LNP023 for the treatment of PNH and C3G.
The pharmacokinetics of nasally administered Foralumab will also be evaluated.
Patient reported outcome to assess clinical responses related to COVID-19 symptoms, as per the FDA guidelines, will also be collected.
FDA-approved oral prescription medicine, 120 mg or 160 mg dependent on weight (<50 kg or ?50 i Continued approval may be contingent upon verification and description of clinical benefit in confirmatory trials. Retevmo may affect both tumor cells and healthy cells, which can result in side effects. Retevmo is an U.S.
Additionally, Regeneron bispecifics are manufactured using similar approaches used for human monoclonal antibody medicines, yielding similar properties and pharmacokinetics. REGN5458 and odronextamab are currently under clinicaldevelopment, and their safety and efficacy have not been evaluated by any regulatory authority.
Submission of an amended protocol to the FDA for the Phase 3 pivotal trial to expand recruitment to approximately 44,000 participants that allows for the enrollment of new populations. The company announced the initiation of its Phase 1b clinical trial to evaluate the safety of a novel investigational therapeutic for COVID-19, PF-07304814.
Researchers must characterize the anti-drug-antibody (ADA) response in preclinical and clinical studies and report any ADA-positive samples as a risk-based approach. Regulatory Considerations for Oligonucleotide Drug Development and Safety In 2024, the U.S.
FDA’s nonprescription advisors find no efficacy for phenylephrine This week, FDA’s Nonprescription Drugs Advisory Committee (NDAC) voted unanimously that current scientific data do not support the efficacy of oral phenylephrine as a nasal decongestant, aligning with FDA analysis — and re-analysis — of data.
FDA solidifies its advice on dose optimization for oncology products Just over a year after it released the draft guidance, the FDA has finalized its guidance on dose optimization for oncology products. The FDA also noted the shifting considerations for evaluating toxicity in modern products meant for long-term or chronic use.
Priothera will use the funds to progress the clinicaldevelopment of mocravimod, a modulator of sphingosine 1 phosphate (S1P) receptors, to enhance the curative potential of allogeneic hematopoietic stem cell transplantation (HSCT) for treating AML. 1. -- -->. -- [if lte IE 8]-->
.
adults age 65 and older.
To encourage the development of such guidance, Congress directed FDA to issue a draft guidance not later than 12 months after the date of FDORA’s enactment and to finalize such guidance not later than 9 months after closing the comment period. intended use population. population in light of the U.S. role in global health.
Patients at a clinical cancer center in Michigan, USA, have been dosed with the experimental drug QN-302, a potential first-in-class novel treatment for advanced or metastatic solid tumors. In January 2023, QN-302 received Orphan Drug Designation by the FDA for the potential treatment of pancreatic cancer.
Podcast : FDA Guidance for Industry Psychedelic Drugs Extensively studied for potential therapeutic efficacy, psychedelic drug development comes with its own set of clinicaldevelopment requirements. Watch the video. Watch it now. The Altascientist : Issue No.
Food and Drug Administration (FDA) Breakthrough Therapy and Fast Track Designations, is now in pivotal testing, and CTP-692 for schizophrenia is currently on track for topline data readout in the first quarter of 2021,” said Roger Tung, Ph.D., CTP-543 for moderate to severe alopecia areata, which received U.S. Canada and Europe.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.









































Let's personalize your content