This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The regulatory environment in Japan for generic drug development is complex and has undergone significant changes in recent years. Regulatory Authority: Pharmaceuticals and Medical Devices Agency (PMDA) The PMDA is the primary regulatory authority responsible for overseeing the drug approval process in Japan.
In a recent survey conducted by ICON, Plc, biomarker selection was identified by 35 percent of respondents as a top challenge among drug developers for phase I trials, second only to navigating regulatory compliance (- 38 percent). To qualify as endpoints, biomarkers used in early phases must be relevant to later stages of drug development.
Mastering Drug Safety: Your Passport to Successful Pharmacology tchichekian Fri, 09/22/2023 - 13:36 HTML Navigating the Depths of Safety Pharmacology Altasciences can help you assess the impact of your therapeutic entity on vital organ systems before first-in-human trials begin.
1] Society and culture Names Eplontersen is the international nonproprietary name. [9] “Population pharmacokinetic/pharmacodynamic modelling of eplontersen, an antisense oligonucleotide in development for transthyretin amyloidosis” British Journal of ClinicalPharmacology. Food and Drug Administration (FDA).
Dr. Löbenberg holds Health Canada licences for research and analytical testing of a wide range of psychedelic compounds under the Controlled Drugs and Substance Act and research and analytical testing licences for cannabis under the Cannabis Act. .
DMT (N,N-dimethyltryptamine).
Prof. Dr. Löbenberg. ” Prof.
CAMBRIDGE, England–( BUSINESS WIRE )– Mission Therapeutics (“Mission”), a drug discovery and development company focused on selectively inhibiting deubiquitylating enzymes (DUBs), has appointed Dr Suhail Nurbhai as Chief Medical Officer (CMO) with immediate effect. 2, 2020 08:00 UTC. This press release features multimedia.
Mode of action The drug acts as an ultra-short-acting 1-selective blocking agent. Contrary to landiolol, exposure to other -blockers such as esmolol amplifies the re-expression of -receptors which explains the drug tolerance effect seen during long-term esmolol infusion. Jump up to: a b “Novel Drug Approvals for 2024” U.S.
FDA Regulations and guidance under OIRA review as of July The White House’s Office of Information and Regulatory Affairs (OIRA) is the regulator of regulators, tasked with ensuring that all federal policies and regulations adhere to laws, existing regulations, federal policies and the wishes of the President.
2] As of July 2022, it is in phase 3 clinical trials for major depressive disorder. [2] 2] Like other kappa opioid antagonists currently under clinical investigation for the treatment of major depression, its efficacy may be compromised by the countervailing activation of pro-inflammatory cytokines in microglia within the CNS. [7]
Biosimilar product developers are expected to conduct foundational analytical studies, animal studies, clinicalpharmacology and immunogenicity assessments, and additional clinical evaluations to demonstrate biosimilarity or interchangeability. Developing biosimilars is an extensive and expensive process. million (IQR: $18-36.7
Perhaps the most important addition to FDA’s agenda is a proposed rule intended to clarify that Laboratory Developed Tests (LDTs) are to be regulated as medical devices under the Federal Food, Drug and Cosmetic Act (FD&C Act). Read our analysis of that rule here and here. ]
December has a huge number of legislative deadlines associated with the Food and Drug Omnibus Reform Act of 2022. Drug Importation: We may start the month of November with greater clarity about the FDA’s thinking about prescription drug importation plans.
Prescription drug advertisements presented through media such as TV and radio must disclose the product’s major side effects and contraindications in what is sometimes called the major statement. This rule also establishes standards for determining whether the major statement in these advertisements is presented in the manner required.
How Improving Diversity Can Benefit Clinical Trials pmjackson Wed, 07/31/2024 - 19:19 In July 2024, the U.S. Food and Drug Administration (FDA) published a draft guidance to ensure greater diversity in clinical trials, which is expected to become a final guidance by June 2025.
By our count, the FDA’s Oncologic Drugs Advisory Committee held seven meetings in 2023; additionally, the Oncology Center of Excellence authored nine new (draft) and six final guidance documents. A key fact about the dostarlimab phase 2 trial for LARC: The drug was stunningly effective. months, according to the GSK presentation.
I am a pharmacist by training and continued with a PhD in ClinicalPharmacology. However, my goal was all the time to work with drug development in the pharma industry, so I moved on and started that journey in 1992 when I took on a role as Clinical Research Manager at AstraHässle, a mid-size Swedish pharma company.
First, an audit of communications and meeting management under the Prescription Drug User Fee Act (PDUFA) Quick background: As AgencyIQ has previously discussed, the FDA collects user fees as part of an essential bargain between regulators and industry. This week, FDA posted several new special notice contract opportunities on SAM.
AgencyIQ has analyzed the agency’s guidance agendas, user fee commitment letters, statutory obligations and international efforts to determine which guidance documents the agency is actively working on this year (and in some cases, in the years ahead as well). Priority A List.
A new era for DTC advertising: On May 20, the FDA’s final rule requiring that direct-to-consumer television and radio advertising contain “clear, conspicuous and neutral” statements regarding the drug’s major side effects and contraindications will go into effect.
Many of those activities are tied to various user fee program commitments; a few others are related to other major pieces of legislation like last year’s Food and Drug Omnibus Reform Act (FDORA). We’re expecting to get a better sense in October about which of these bills might eventually start to move forward and potentially become law.
December also has a huge number of legislative deadlines associated with the Food and Drug Omnibus Reform Act of 2022. Drug Importation: We may soon receive greater clarity about the FDA’s thinking about prescription drug importation plans. December also typically brings end-of-year perspectives on the agency’s accomplishments.
The documents describe all the guidance documents that are under development and that may be published in a given year. Note: If a link no longer works, it is likely because OIRA has since cleared the document.)
On March 26, the Supreme Court will hear the case of Alliance for Hippocratic Medicine vs. FDA , a case which calls into question FDA’s authority to make regulatory changes, to be free from unreasonable scrutiny, and to ensure that the abortion drug mifepristone remains both approved and widely available.
The oncology drug development landscape is evolving rapidly, driven by the deployment of targeted therapies in precision medicine and regulatory initiatives like the FDAs Project Optimus. It also covers strategies for drug developers who have yet to identify a biomarker, helping them advance their programs effectively.
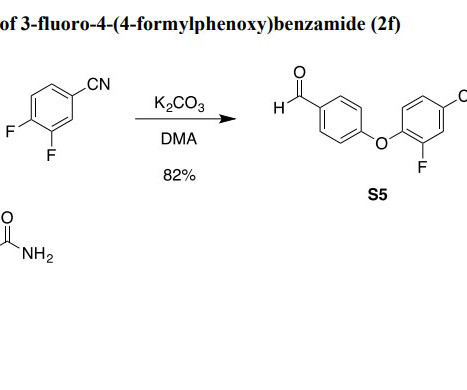
The DMSO solution of the alkyl bromide were slowly transferred in order to maintaining an internal temperature of 55.05 Methanol is then removed by vacuum distillation at an internal temperature of NMT 35 C. 20] Acoramidis is the international nonproprietary name. [21] Food and Drug Administration. 26 November 2024. .
A closer look at the seven new proposed regulations Stage of Rulemaking Title Estimated Publication Synopsis Proposed Rule Amendments to the Current Good Manufacturing Practice Regulations for Drug Products February 2025 FDA is proposing to amend the Current Good Manufacturing Practice Regulations for Drug Products.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.






























Let's personalize your content