This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
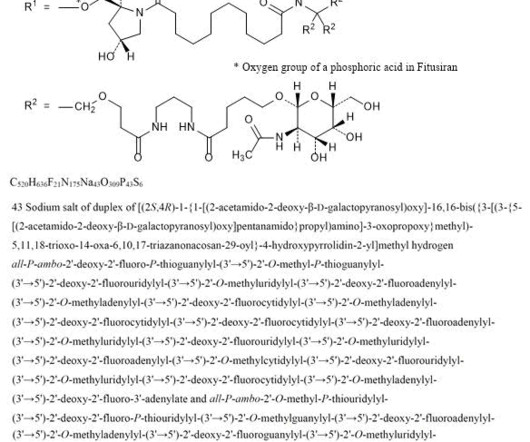
2] Fitusiran was approved for medical use in the United States in March 2025. [2] 2] History The efficacy and safety of fitusiran were assessed in two multicenter, randomized clinicaltrials which enrolled a total of 177 adult and pediatric male participants with either hemophilia A or hemophilia B. [2] Fitusiran 1711.0g/mol,
Pfizer and BioNTech initiated the BLA by submitting the nonclinical and clinical data needed to support licensure of the COVID-19 vaccine for use in individuals 16 years of age and older. The Pfizer-BioNTech COVID-19 Vaccine has not been approved or licensed by the U.S.
FDAApproves Danyelza (naxitamab-gqgk) for the Treatment of Neuroblastoma. It’s very exciting to see this treatment go from being an experimental therapy used at my daughter’s bedside to now being FDAapproved. The FDA granted approval under the accelerated approval regulation. NEW YORK, Nov.
During the meeting, the FDA provided encouraging feedback regarding the Phase 3 study of omidubicel pertaining to the pre-specified primary and secondary endpoints. The FDA also recommended that Gamida Cell generate additional manufacturing-related data prior to requesting a pre-Biologics License Application (BLA) meeting.
We will now rapidly move to initiate our Phase 2 trial of Berubicin for adults with GBM and expect to begin enrolling patients in the first quarter of next year,” commented John Climaco , CEO of CNS Pharmaceuticals. and 2 trials planned by our sublicensee WPD in Poland.
1] [2] It was developed by Vertex Pharmaceuticals , [5] and was approved for medical use in the United States in January 2025. [2] 2] [6] Suzetrigine is the first medication to be approved by the US Food and Drug Administration (FDA) in this new class of pain management medicines. [2] Food and Drug Administration (FDA).
Fast Track designation is well-timed, as we anticipate starting our Phase 2 clinicaltrial in hospitalized COVID-19 patients this month, and should help bring Brilacidin to patients faster in these dire times.”. Brilacidin for UP/UPS was licensed to Alfasigma S.p.A.
Phase III BRIDGE open-label, switch-over clinicaltrial met key objectives for safety and efficacy.
” In May 2020 , Protalix and Chiesi Global Rare Diseases announced the submission of a Biologics License Application (BLA) to the U.S. Protalix has licensed to Pfizer Inc.
2] Crinecerfont was approved for medical use in the United States in December 2024. [2] 2] [3] The US Food and Drug Administration (FDA) considers it to be a first-in-class medication. [4] 2] In the first trial, 122 adults received crinecerfont twice daily and 60 received placebo twice daily for 24 weeks. [2] 1 December 2024.
2] Vorasidenib was approved for medical use in the United States in August 2024. [2] 2] [3] It is the first approval by the US Food and Drug Administration (FDA) of a systemic therapy for people with grade 2 astrocytoma or oligodendroglioma with a susceptible isocitrate dehydrogenase-1 or isocitrate dehydrogenase-2 mutation. [2]
“Population pharmacokinetic/pharmacodynamic modelling of eplontersen, an antisense oligonucleotide in development for transthyretin amyloidosis” British Journal of Clinical Pharmacology. S2CID 250989659. ^ “Eplontersen: FDA-Approved Drugs” U.S. Food and Drug Administration (FDA). doi : 10.1111/bcp.15468.
If approved, Actemra/RoActemra would be the first U.S. FDAapproval is expected in the second half of this year. About the Actemra®/RoActemra® (tocilizumab) COVID-19 ClinicalTrial Programme Roche’s clinicaltrial programme evaluated the safety and efficacy of Actemra/RoActemra in hospitalised patients with COVID-19.
1, 2020 /PRNewswire/ — Sosei Group Corporation (“the Company”) (TSE: 4565) announces it has entered into a global collaboration and license agreement with Biohaven Pharmaceutical Holding Company Ltd. (“Biohaven”, NYSE: BHVN). .
TOKYO and CAMBRIDGE, England , Dec.
FDA 12/1/2022, To treat adults with relapsed or refractory acute myeloid leukemia with a susceptible isocitrate dehydrogenase-1 (IDH1) mutation, Rezlidhia Olutasidenib , sold under the brand name Rezlidhia , is an anticancer medication used to treat relapsed or refractory acute myeloid leukemia. [1] 1] It is taken by mouth. [1] Hz, 1 H), 4.62−4.75
Landiolol 133242-30-5 ONO-1101 Ono 1101 WHO 7516 FDAAPPROVED 11/22/2024, Rapiblyk , To treat supraventricular tachycardia C25H39N3O8 509.6 Randomized clinicaltrials have been published to compare landiolol with placebo< [21] [22] [23] diltiazem, [24] and amiodaron [25] in patients with or without heart failure.
Pfizer plans to file for full FDAapproval of Covid vaccine at the end of this month ( CNBC ).
The FDA is set to authorize the Pfizer-BioNTech vaccine for those 12-15 years old by early next week.
Big three drug distributors blame doctors, regulators in trial over opioid epidemic ( Reuters ).
Food and Drug Administration (FDA) has extended the Prescription Drug User Fee Act (PDUFA) date for review of the Company’s Biologics License Application (BLA) seeking accelerated approval of pegunigalsidase alfa (PRX–102) for the proposed treatment of adult patients with Fabry disease. Protalix has licensed to Pfizer Inc.
2 , 3 Lazertinib was first approved in South Korea on January 18, 2021, for the treatment of EGFR T790M mutation-positive non-small cell lung cancer (NSCLC) with EGFR mutations. 1 It was approved by the FDA on August 19, 2024. 1 It was approved by the FDA on August 19, 2024. Food and Drug Administration (FDA).
1] Motixafortide was approved for medical use in the United States in September 2023. [2] 4 Similar in mechanism to the previously approved plerixafor , motixafortide is an inhibitor of C-X-C Motif Chemokine Receptor 4 (CXCR4), a protein that helps to anchor stem cells to bone marrow matrix. 1] It is given by subcutaneous injection. [1]
Jump up to: a b c d e f g h “FDAapproves new drug for hypoparathyroidism, a rare disorder” U.S. Food and Drug Administration (FDA) (Press release). Text was copied from this source which is copyright European Medicines Agency. Reproduction is authorized provided the source is acknowledged. 9 August 2024.
Pfizer and BioNTech expect to file a Biologics License Application for possible full regulatory approval in 2021.
Food and Drug Administration (FDA) has authorized the emergency use of the mRNA vaccine, BNT162b2, against COVID-19 in individuals 16 years of age or older. About the Phase 2/3 Study.
Pfizer and BioNTech have submitted Phase 1 data – part of their Phase 1/2/3 clinicaltrial program – evaluating the safety, tolerability, and immunogenicity of a third dose of the COVID-19 vaccine in U.S. adult participants from the Phase 1 trial of the two-dose series. In the U.S.,
based contract manufacturing business, Benuvia Manufacturing, which has significant chemistry and formulation capabilities, including manufacturing our FDA-approved cannabinoid drug, SYNDROS ® ,” said Todd C. ” Clinical Highlights. We look forward to supporting Radius through our U.S. Davis, executive chairman of Benuvia.
The new findings from the Phase 3 clinicaltrials (ADvocate 1 and 2) showed eight out of ten patients who achieved clinical response (EASI-75*) with lebrikizumab monotherapy at 16 weeks maintained skin clearance at one year of treatment with the once every two weeks or four weeks regimen. . Almirall S.A.’s Almirall S.A.’s
Retrieved 10 December 2023. ^ “Novartis receives FDAapproval for Fabhalta (iptacopan), offering superior hemoglobin improvement in the absence of transfusions as the first oral monotherapy for adults with PNH” Novartis (Press release). 5 December 2023. . twitter +919321316780 call whatsaapp EMAIL. 5 December 2023.
SetPoint Medical received FDA Investigational Device Exemption (IDE) approval for a multicenter, double-blind, randomized, sham-controlled pivotal trial that will enroll up to 250 patients at 40 clinicaltrial sites in the U.S. Small Molecule Inhibitors. Anti-CD40L Antibody.
Molecular Weight: 631.700 FDAAPPROVED, To treat moderately to severely active ulcerative colitis in adults, 10/12/2023 Velsipity Etrasimod , sold under the brand name Velsipity , is a medication that is used for the treatment of ulcerative colitis (UC). [1] “FDAApproves New Drug for Ulcerative Colitis” Medscape.
3] In November 2023, capivasertib was approved in the United States for people with hormone receptor-positive, human epidermal growth factor receptor 2 -negative breast cancer when used in combination with fulvestrant. [3] Jump up to: a b c d e f “FDAapproves capivasertib with fulvestrant for breast cancer” U.S.
Scott and colleagues focused on six genes that encode potential drug targets licensed or in development by GlaxoSmithKline for the treatment of obesity or diabetes. That’s critical considering that just 1 in 10 drug candidates entering human clinicaltrials successfully goes on to receive FDAapproval [5].
Pfizer and BioNTech are extremely grateful to the study volunteers and investigative site staff in the clinicaltrial program, as their involvement was crucial to today’s important milestone in the companies’ efforts to address the COVID-19 global pandemic. as well as Europe, Latin America, and South Africa. About the Study.
Food and Drug Administration (FDA) has accepted for review the supplemental Biologics License Application (sBLA) for Dupixent ® (dupilumab) as an add-on treatment for children aged 6 to 11 years with uncontrolled moderate-to-severe asthma. In children younger than 12 years of age, Dupixent should be administered by a caregiver.
The positive opinion adopted by the CHMP is based on an evaluation of interim safety and efficacy data from a clinicaltrial of a booster dose of the vaccine in those aged 16 and over, together with published literature and post authorisation data plus real-world evidence from the use of booster doses in young patients in Israel.
Food and Drug Administration (FDA) approval for patients with locally advanced or metastatic NSCLC with epidermal growth factor receptor (EGFR) exon 20 insertion mutations, based on data showing an ORR of 40 percent (95 percent CI, 29 – 51) and median duration of response of 11.1 months (95 percent CI, 6.9 – NE). [7].
MRNA-1273 (SARS-CoV-2 vaccine) FDAApproval History. These forward-looking statements are based on Moderna’s current expectations and speak only as of the date hereof. Source: Moderna, Inc. . Posted: January 2021. Source link.
BioAge is on the cusp of taking pilot therapies BGE-117 and BGE-175 into clinicaltrials, targeting the first half of 2021. Pear’s reSET, reSET-O and Somryst are the first PDTs to receive FDAapproval for treating disease.
If REGN-COV2 proves safe and effective in clinicaltrials and regulatory approvals are granted, Regeneron will manufacture and distribute it in the U.S. Department of Health and Human Services under OT number: HHSO100201700020C. Regeneron has partnered with Roche to increase the global supply of REGN-COV2 beginning in 2021.
MRNA-1273 (SARS-CoV-2 vaccine) FDAApproval History. These forward-looking statements are based on Moderna’s current expectations and speak only as of the date hereof. Source: Moderna, Inc. . Posted: January 2021. Source link.
A surrogate endpoint is a marker, such as a laboratory measurement, radiographic image, physical sign or other measure that is thought to predict clinical benefit but is not itself a measure of clinical benefit. The use of a surrogate endpoint can considerably shorten the time required prior to receiving FDAapproval.
Food and Drug Administration (FDA) planned for soon after the required safety milestone is achieved, which is currently expected to occur in the third week of November.
We will continue to collect further data as the trial continues to enroll for a final analysis planned when a total of 164 confirmed COVID-19 cases have accrued.
MRNA-1273 (SARS-CoV-2 vaccine) FDAApproval History. These forward-looking statements are based on Moderna’s current expectations and speak only as of the date hereof. Source: Moderna, Inc. . Posted: December 2020. Source link.
Data from the Phase 1/2/3 clinicaltrial supported an Emergency Use Authorization for casirivimab and imdevimab administered together, granted by the U.S. If the therapy proves safe and effective in clinicaltrials and regulatory approvals are granted, Regeneron will manufacture and distribute it in the U.S. ,
We remain focused on continuing to serve patients, protecting the health and safety of our employees and the communities in which we live and work, and supporting patients in clinicaltrials. The COVID-19 impact has varied by study and program, but there has been little timing impact on fully-enrolled trials.
The sponsor is the pharmaceutical company conducting the trial. If you mean using a different contract research organization (CRO) for the different phases of clinicaltrials – that’s different. The sponsor-initiated IND is conducted by a pharma company seeking commercial approval. There is no problem doing this.
Results showed no significant difference in virologic or clinical efficacy between the REGN-COV2 high dose (8 grams) and low dose (2.4 Based on this finding, Regeneron is reviewing potential changes to dosing in the ongoing outpatient clinicaltrial given the current limited supply of REGN-COV2.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.












































Let's personalize your content