This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Artificial intelligence (AI) has revolutionised many industries, yet its adoption in pharmaceutical drugdevelopment has been notably slower. For years, AI and machine learning (ML) were often dismissed as little more than advanced statistics with little practical value in drugdevelopment.
While there is no universally accepted definition for rare disease, it is estimated that more than 10,000 distinct rare conditions exist, and people living with these rare diseases represent as much as 10% of the global population. In rare diseases, evidence gaps are often significant, necessitating de novo evidence generation.
For example, transcriptomic processes are showing the potential to identify and track failures in gene expression and gene regulation of amyloid and tau-related biomarkers, understood as precursors to the onset of Alzheimers disease (AD).
Traditional risk managers, by their job definition, are highly cautious of the result sets provided by the analytics teams. Banks have silos, these silos have been created due to mergers, regulations, entities, risk types, chinese walls, data protection, land laws or sometimes just technological challenges over time.
Establish a single source of truth Create a glossary that doesn’t read like a legal document Accept that these definitions will change more often than a teenager’s social media profile It’s not perfect, but it’s governance, not a philosophical treatise on the nature of reality. So very, very wrong.
Data privacy has become an increasingly complex subject, especially with the introduction of the California Consumer Privacy Act (CCPA) and similar regulations emerging in other states. Non-compliance with these regulations can result in hefty fines, as seen with Sephora’s recent $1.2
The company said it continues to expand its manufacturing capacity for the therapy as it closes in on a third approved indication, with seven facilities across four continents contributing to the production of the drug worldwide. “We Matt Fellows. Source link.
Tobolowsky & Véronique Li, Senior Medical Device Regulation Expert & David B. The draft guidance therefore continues to “recommend” sponsors go beyond what is required in its regulations in its communications to FDA based on DMC recommendations, highlighting the tension between what FDA needs and what it wants.
Early and Ongoing Engagement Will Save Effort During Your Oncology Clinical Trial Early and ongoing engagement with regulatory agencies is invaluable; initiating dialogue early in the development process facilitates a clearer understanding of regulatory requirements and expectations and builds a relationship with regulators.
By Riëtte van Laack — The dietary supplement exclusionary clause is, as its name suggests, a clause in the Federal Food, Drug, and Cosmetic Act (FDC Act) definition of dietary supplement. That clause excludes those ingredients that were first marketed as drug ingredients. What led to NPA’s lawsuit?
Regulatory affairs workers get to tell a story about a drug to the regulators and – keeping the end in mind, which is the drug label or prescribing information – is a good way to communicate with regulators. Every drug has a story: an end game. By definition, this is the cutting edge, and we are in it.
The Folly of Following Trends Trends are, by definition, people following a current style or popular approach. At that time, their industry was driven by government regulations and tariffs. Not once have I had a group who wasn’t able to produce an intriguing idea within five to ten minutes based on a random word. Great ideas abound!
Before I begin, I just want to caveat everything with the fact that HIPAA is a complex regulation open to interpretation, and in the end your legal and compliance teams need to be comfortable with how you handle data and the risk associated with those methods. The other part of the definition is what constitutes “health information.”
Understanding Adaptive and Assistive Technologies Adaptive Technologies: Definition: Adaptive technologies are specialized tools and modifications designed to assist individuals with disabilities in performing tasks that might otherwise be challenging or impossible. These technologies often support independence and improve quality of life.
Key Takeaway RWD and RWE play an important role in rare diseases, where capturing relevant data to support drugdevelopment is challenging. To learn more about using RWD and RWE in rare disease programs, read our guide on Using Real-World Data and Real-World Evidence to Support Rare Disease Programs.
BY LAURA DIANGELO, MPH | JUL 10, 2024 3:47 PM CDT Regulatory background: The FDA is responsible for overseeing information about regulated products. The FDA is responsible for ensuring that medical products are adequately labeled in accordance with federal regulations, including the product’s “intended use” and relevant safety information.
Two weeks ago, FDA published a draft of its latest drugdevelopment guidance explaining how drug and biological product developers can use this pathway to meet the statutory standard for efficacy. The guidance’s second warning reminds drugdevelopers that FDA regulations (i.e.,
While the agency has been working to implement its rule, which has broad-reaching impacts for diagnostics and drugdevelopers, other moves by the Supreme Court and legislators could impact the agency’s next steps. AgencyIQ provides a status update for regulated industry. diagnostic products are regulated as medical devices.
Today, the FDA unveiled a contract notice that explains the process for how it will develop that report, obtain public input, and deliver the report’s final recommendations. a disease or condition is defined as rare by the Orphan Drug Act (ODA) if it affects fewer than 200,000 people. regulators. In the U.S., Recently, U.S.
Diagnostics landscape analysis: Key issues and a look ahead at 2024 Diagnostic regulation in the U.S. Following significant policy proposals in recent years, 2024 is gearing up to be a profoundly impactful year for diagnostics developers and the policymakers who regulate them. is a constantly evolving topic. In the U.S.,
In particular – potential issues with applying the policy to practice, the role of different stakeholders and the new proposed definitions. IVDs are regulated as medical devices in the U.S., However, this definition presents many questions. but since the 1970s, the FDA has expressed “enforcement discretion” (i.e.,
In addition to describing the intended purpose of the procedure, the ATP outlines anticipated performance criteria and Critical Quality Attributes (CQAs), which facilitate technology selection, procedure design and development, performance monitoring, and continual improvement.
Even the definition of “good” is harder to define with unstructured data than structured data. Conclusion Enterprise data, particular in regulated industries, rely on robust, effective and ubiquitous quality pipelines. Working with unstructured data, particularly at scale, can be difficult. An elusive goal, at best.
The standards would, as stated previously, be voluntary, unless mandated by statute or regulation, and they cannot conflict with existing law or regulation. Consensus does not require unanimity, but a general agreement. Existing published VCS may be identified internally by FDA or externally by stakeholders.
What will the orphan drug market exclusivity haircut mean for industry? Just over a month ago, the European Commission released its proposal for the new pharmaceutical directive and regulation. Other orphan drugs, which, by definition, address an unmet medical need, “only” receive 9 years of market exclusivity.
Key stakeholders weigh in on FDA’s Laboratory Developed Test Rule. The 60-day comment period on the FDA’s proposed rule to revamp the way that laboratory developed tests (LDTs) are regulated ended on December 4th, 2023. While the FDA has traditionally regulated diagnostic products as medical devices in the U.S.,
Security and Compliance: ChatBOTs prioritize security and compliance by implementing measures such as encryption, access controls, and adherence to data protection regulations to safeguard user information and ensure privacy. Let’s add a pipeline, Definition will be Pipeline Script. You must add this inside job like the below image.
The legislation further explained that for a certain method to qualify as a “platform technology,” it must demonstrate “reasonable likelihood to bring significant efficiencies to drugdevelopment and review processes.” How can sponsors “prove” that these three criteria are met?
Compliance and Regulation: Azure AD B2C provides compliance with industry regulations such as GDPR and HIPAA, as well as support for authentication standards like OpenID Connect and OAuth 2.0. This makes it easier for businesses to comply with regulatory requirements and ensure the security of user data.
However, the new lengthy regulation raised many questions by the industry, and Perficient’s Financial Services Risk and Regulatory consultants have been researching answers and questioning regulators to provide in order to provide our current and future clients the answers to the following questions that have been raised.
The general notion is that patients should be viewed individually, rather than strictly as members of some larger general population, and that their specific genetic background, environment, and lifestyle choices should be considered throughout drugdevelopment to the point of treatment and continuing patient care.
The work could represent early steps toward modernizing one part of FDA’s regulation of this complicated product class. BY AMANDA CONTI | JAN 16, 2024 7:43 PM CST Regulatory background: complex generics Unlike new drug products, generic drugs do not require extensive clinical evidence to demonstrate safety or efficacy.
For example, in September 2022 the FDA finalized a guidance document on how sponsors of drug product submissions could effectively flag RWE for regulators. Overall, the FDA urged developers to engage with the agency early – including through a Type C meeting – to go over their planned protocol and statistical analysis plan (SAP).
During the development cycle of a regulated therapeutic, the transfer of the manufacturing process is inevitable. The process will be transferred from the development lab to a pilot or small-scale manufacturing facility and, if the program is successful, to a facility for commercial manufacturing.
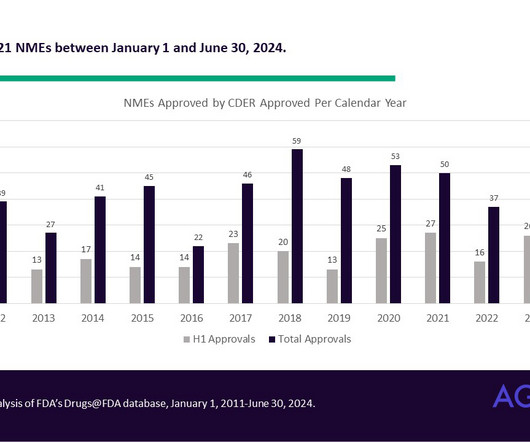
If a drug was previously approved for cancer, but is now approved for a cardiology condition, it would not be considered novel. While the definition of NME has changed over the years, it can sometimes include a combination product consisting of at least one drug that has previously been approved.
Because CDx are diagnostic devices, they are not considered part of a combination product but are regulated separately as medical devices. But not all tests and diagnostics used with a drug product are actually CDx.
FDA’s Laboratory Developed Test (LDT) Rule Quick background: The FDA recently published a new final rule to formally extend regulatory oversight over laboratory developed tests (LDTs), which the agency has long maintained are a subset of in vitro diagnostics (IVDs) and therefore should be regulated as a medical device in the U.S.
In 1972, when regulatory authority for biologic products was transferred from the National Institutes of Health (NIH) to the FDA, the agency extended the two-study framework to biologic regulation as well. The Heckler case helped to refine the definition of “substantial evidence” in two important ways.
The FDA also implemented new flexibilities for certain regulated products and processes, typically via enforcement discretion. As before, the guidance provides recommendations on drugdevelopment for COVID-19 across five domains: population, trial design, efficacy endpoints, safety considerations and statistical considerations.
BY RACHEL COE, MSC JUN 6, 2023 5:00 PM CDT What are nonclinical studies and when are they conducted in drugdevelopment? It also contains a definition of the term immunotoxicity— the unintended immunosuppression or stimulation (including hypersensitivity) —which was notably absent from the draft.
Modeled on the Operation Warp Speed vaccine development communications approach, the pilot aims to test out a rapid communications approach for accelerating rare disease drugdevelopment. The idea is that the development challenges faced for a disease which only affects a few hundred patients in the U.S.
Previously, burdensome side effects may have been written off as a necessary part of treatment and benefits measured in additional weeks or months of survival time; today, though, patients, clinicians, and regulators expect more.
The EMA started providing guidance on the clinical development of anticancer therapeutics in 1996. The document has been updated over time to implement advances in understanding cancer and drugdevelopment. In 2005, the guidance was updated to include non-cytotoxic drugs were gaining in importance.
The regulations also explain that this trait can be demonstrated via appropriate laboratory tests or adequately controlled clinical data. The first (pages 1-5) explains what regenerative medicine products are, provides an overview of what “risk management” means, and cites the requirements that drugdevelopers must satisfy per the U.S.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.













































Let's personalize your content