This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
This of course, make sense—after decades of experience implementing the Hatch-Waxman, Congress and FDA had learned a few new tricks by 2009/2010. Amongst other things, FDA co-opted many of the same definitions for key terms for implementation of the BPCIA. FDA borrowed this definition from 21 C.F.R.
Kymriah was preliminarily granted orphan medicinal product designation by the European Commission (EC) forFL.However, Kymriah would have the occasion to present an important treatment option for those cases with r/ r FL in need of potentially definitive issues, If approved in this implicit third suggestion.
The draft guidance provided language stating that all products, whether biosimilar or interchangeable, be referred to as “biosimilar” on the product label, with a general definition explaining the term biosimilar. Today, FDA published a revised version of the Q&A guidance on promotional labeling. Yes, the guidance confirms.
Radius” or the “Company”) (Nasdaq: RDUS) today announced a definitive agreement to acquire the global development and commercialization rights to Benuvia Therapeutics Inc. ’s PWS is an orphan disease with major endocrine and behavioral manifestations and no FDA-approved therapies for the treatment of hyperphagia.
The sponsor-initiated IND is conducted by a pharma company seeking commercial approval. Q: When you want to do a study of an FDA-approved drug under an investigational indication, is an IND still required or can you seek an exemption? Q: What is the definition of a drug? Then you can submit an application for approval.
These follow-on data provided the first definitive prospective evidence demonstrating anti-viral activity for a treatment regimen now available for COVID-19, and also further documented the ability of this treatment to decrease the need for further medical attention,” said George D. Have chronic kidney disease. Have diabetes.
Today’s data, involving an additional 524 patients from the ongoing Phase 2/3 trial, provides definitive final virology results and meets the clinical endpoint of reducing medical visits. TARRYTOWN, N.Y. , 28, 2020 /PRNewswire/ — . Regeneron has shared these results with the U.S.
1 Sairiyo has an exclusive license from a research and development organization to develop and commercialize reformulated Cepharanthine for all diseases and exclusive rights to the patent, method of manufacturing, clinical supply, pre-clinical data and know-how to support FDA clinical trials. PharmaDrug Inc.
The mechanism of action for mepolizumab has not been definitively established. It is not currently approved for use in COPD anywhere in the world. The following information is based on the US Prescribing Information for Nucala in licensed indications only. Important safety information. CONTRAINDICATIONS.
The mostly redacted, then publicly available letter accompanying the basis for the recommendation stated that FDA was making a recommendation about marijuana, referring to botanical cannabis ( Cannabis sativa L.) within the CSA’s definition of “marihuana” or “marijuana” based on FDA’s eight factor analysis. 21 U.S.C. §
For this reason, the guidance advises sponsors that “some technologies that industry and FDA have historically considered to be platform technologies might not meet the statutory definition and statutory eligibility factors and, if not, would not be eligible for the designation program.” which would never be acceptable.
All products are received in one of two forms: A New Drug Application (or NDA, for pharmaceuticals) or a Biologics License Application (BLA, for biologics). Novel drug products are defined as products that have never been approved for any indication. The FDA also refers to novel products as “New Molecular Entities,” or NMEs.
Casirivimab and imdevimab injection is an investigational combination therapy and has been authorized by FDA for the emergency use described above. Casirivimab and imdevimab injection is not FDAapproved for any use. Definition of High-Risk Patients. Have chronic kidney disease. Have diabetes.
0910-AI26 September 2023 March 2023 Final Rule Stage Biologics License Applications and Master Files The Biologics Price Competition and Innovation Act of 2009 (BPCI Act) deemed any biological product approved in a new drug application (NDA) to be a biologics license application (BLA) on March 23, 2020. fit in this definition.
The FDA grants Emergency Use Authorization to medicines that may help diagnose, treat or prevent a life-threatening disease when adequate and approved alternatives are not available. The EUA is temporary and does not take the place of a formal biologics license application (BLA) submission review and approval process.
The FDA grants Emergency Use Authorization to medicines that may help diagnose, treat or prevent a life-threatening disease when adequate and approved alternatives are not available. The EUA is temporary and does not take the place of a formal biologics license application (BLA) submission review and approval process.
Food and Drug Administration (FDA) in high-risk patients who have confirmed COVID-19 but are not currently hospitalized. The EUA is temporary and does not take the place of a formal biologics license application (BLA) submission review and approval process and the use of the antibody cocktail remains investigational.
Curiously, there is no direction for the patient to seek out the full FDA-approved labeling. Information included in the PMI : FDA intends that the PMI would “highlight the most important information that patients need to know to help them use their prescription drug products safely and effectively.”
See “Reconciliation of Non-GAAP Measures” for a definition of the terms and reconciliation tables.).
A follow-on procurement contract with HHS is expected for the delivery of raxibacumab, the Company’s Food and Drug Administration-approved anthrax monoclonal antibody therapeutic, to the Strategic National Stockpile (SNS).
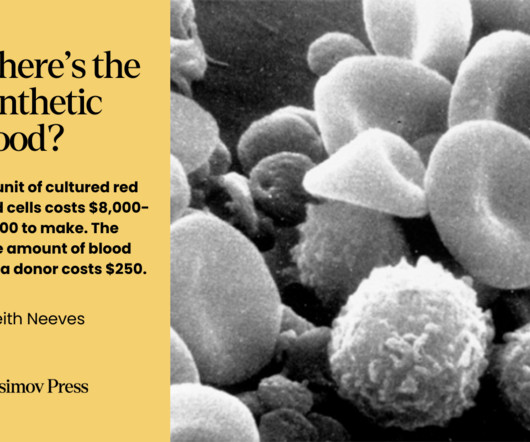
While blood doesn’t fit neatly into the various definitions of an organ , it still comprises a discrete system with specific functions. Despite the initial promise of PFCs and FDAapproval of a product called Fluosol-DA in 1989, PFCs have many drawbacks.
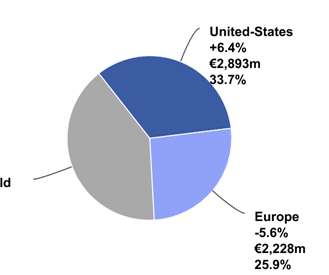
Changes in net sales are expressed at constant exchange rates (CER) unless otherwise indicated (definition in Appendix 7) (1) In order to facilitate an understanding of operational performance, Sanofi comments on the business net income statement. Business net income is a non-GAAP financial measure (definition in Appendix 7).
FDA analyzed anorexia related to a medical condition, anxiety, epilepsy, inflammatory bowel disease, chemotherapy-induced nausea and vomiting, pain, and post-traumatic stress disorder. The proposed rescheduling would not apply to synthetically derived THC outside of the CSA definition of marijuana, which would remain in schedule I.
So what about all of the “biotic” terms that pop up in the news, on products labels and even in FDA warning letters? Unfortunately, there is no regulatory body that governs the terminology used in this space, although there are definitions that are generally accepted by the scientific community.
January 2024 Amendments to Patent Term Restoration (Proposed Rule) FDA is proposing to amend its Patent Term Restoration regulations to identify the effective date of approval for scheduled drugs, to define when the 60-day period referred to in 35 U.S.C. fit in this definition.
The Complaint further alleges that data collected and submitted in support of full (as opposed to emergency use) FDAapproval demonstrated the misleading nature of earlier statements. The FDA, however, did not and does not share that belief. FDA (8/23/21) press release (emphasis original). Health & Safety C.
Before a biologic may be marketed, the manufacturer must obtain a license from the FDA. Approval of an application “constitute[s] a determination” by the FDA “that … the product meet[s] applicable requirements to ensure the continued safety … of such products.” See 42 U.S.C. § See 21 C.F.R. 2022 WL 4305656, at *7.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.






























Let's personalize your content