This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
FTC, deep in its foray into the Orange Book, filed an Amicus Brief in the case arguing that the patents do not claim any FDA-approved drug. While the relevant regulation defines “drug product” as the “finished dosage form,” which theoretically includes the delivery device, the Court again falls back on the wording in 21 C.F.R.
Completion of the transaction is subject to customary closing conditions, including completion of due diligence, negotiation of definitive agreements and receipt of all necessary approvals. Persons”, as such term is defined in Regulations under the U.S.
By Riëtte van Laack — For those readers unfamiliar with the regulation of animal food ingredients in the United States, below is a brief background. In the United States, animal food (feed) regulations are enforced by state and federal regulatory officials. AAFCO develops model regulations, that serve as model for the states.
This of course, make sense—after decades of experience implementing the Hatch-Waxman, Congress and FDA had learned a few new tricks by 2009/2010. Amongst other things, FDA co-opted many of the same definitions for key terms for implementation of the BPCIA. FDA borrowed this definition from 21 C.F.R.
Gaulkin — In May 2023, we posted about a CMS proposed regulation that sought to make a wide variety of changes to the Medicaid Drug Rebate Program (MDRP), including a new “price verification survey,” and a controversial proposal to require “stacking” of discounts to different customers when determining best price.
The mostly redacted, then publicly available letter accompanying the basis for the recommendation stated that FDA was making a recommendation about marijuana, referring to botanical cannabis ( Cannabis sativa L.) within the CSA’s definition of “marihuana” or “marijuana” based on FDA’s eight factor analysis. 21 U.S.C. §
Food and Drug Administration (FDA)-approved therapies for treating PAH were primarily vasodilators, designed to overcome the imbalance between vasoactive and vasodilator mediators and to restore endothelial cell function. Their early definition — as well as plans for recording and tracking — is a major factor in a trial’s success.
Our objective is to work with Bruker and our clinical partners to deliver an effective and high throughput assay protocol, that can be applied in the clinical setting, with the required sensitivity and specificity and CE/FDAapprovals to provide a useful additional diagnostic tool in the fight against the coronavirus.
The 51 regulations that FDA is currently working on The FDA today unveiled its much-anticipated Spring 2023 Unified Agenda, a document outlining the regulations the agency plans to release in 2023 and beyond. The anticipated date of publication is June 2023, meaning we should see this regulation imminently.
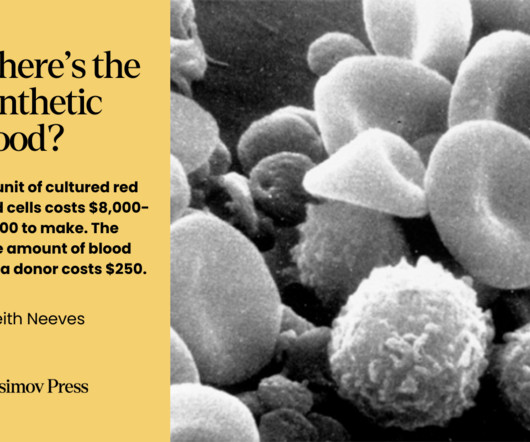
While blood doesn’t fit neatly into the various definitions of an organ , it still comprises a discrete system with specific functions. Despite the initial promise of PFCs and FDAapproval of a product called Fluosol-DA in 1989, PFCs have many drawbacks. It can be frozen or freeze dried.
The treatment had been granted breakthrough therapy designation, but FDAapproval would ultimately rest on the sponsor’s ability to demonstrate clinically meaningful improvement. Key Takeaway RWD and RWE play an important role in rare diseases, where capturing relevant data to support drug development is challenging.
In a new final rule, FDA carves out a regulatory niche for medical gases Industry has been lobbying FDA and Congress to regulate medical gases different from other types of drug products since the 1970s. Following this process, FDASIA directed the FDA to submit a report to Congress summarizing its findings.
Radius” or the “Company”) (Nasdaq: RDUS) today announced a definitive agreement to acquire the global development and commercialization rights to Benuvia Therapeutics Inc. ’s PWS is an orphan disease with major endocrine and behavioral manifestations and no FDA-approved therapies for the treatment of hyperphagia.
The standard for the audit used depends on the type of vendor and would include the applicable regulations and ICH guidelines. The sponsor-initiated IND is conducted by a pharma company seeking commercial approval. Q: What is the definition of a drug? A: For FDA 1572 is a Statement of Investigator under US Regulations/Laws.
This year we completed investigational new drug (IND) enabling studies of ONC-841, which is an antagonist of Siglec-10, and received US Food and Drug Administration (FDA) approval for first-in-human study (FIH) in patients with solid tumours. The first patient has been dosed in the last quarter.
PROs, included in clinical trials as primary or secondary endpoints , are increasingly recognized by regulators, clinicians, and patients as valuable tools. Role of PROs in drug approvals A recent review sought to comprehensively characterize the inclusion of PROs and regulatory considerations in FDA-approved novel oncology drugs.
BY CHELSEY MCINTYRE, PHARMD | JUN 3, 2024 8:43 PM CDT Regulatory background: DSHEA and dietary supplements The Dietary Supplement Health and Education Act ( DSHEA ) of 1994 defines the FDA’s authority in the regulation of dietary supplement products and dietary ingredients. See AgencyIQ’s analysis of the draft guidance here. ]
So what about all of the “biotic” terms that pop up in the news, on products labels and even in FDA warning letters? Unfortunately, there is no regulatory body that governs the terminology used in this space, although there are definitions that are generally accepted by the scientific community.
FDAapproval of a new drug for the condition under study) Impact of participation on alternative therapies (e.g., IRB Review of Minor Study Changes Federal regulations and guidance do not provide a singular definition for “minor” changes to research. a data breach) Decrease in expected benefits to participation (e.g.,
Following FDAapproval, these products are granted seven years of marketing exclusivity, preventing FDA from approving the “same drug for the same disease or condition” if the applicant does not have the right to reference the product (i.e., DORIS MATSUI (D-Calif.) and GUS BILIRAKIS (R-Fla.), TAMMY BALDWIN (D-Wisc.).
The FDA recently concluded its work on a proposed rule focused on PMI. The regulator sent the rule to the White House’s Office of Information and Regulatory Affairs (OIRA) on October 4, 2022. The content of the PMI : The regulation describes, in broad terms, what must be included in the PMI.
The delayed enforcement is part of FDA’s finalized guidance, “Compliance Policy for Cosmetic Product Facility Registration and Cosmetic Product Listing.” NOV 8, 2023 10:02 PM CST Regulatory background The Federal Food, Drug, and Cosmetics Act (FD&C Act) is administered by the Food and Drug Administration (FDA) and regulates cosmetics.
For context, while prescription drug products are required to comply with FDA’s regulatory requirements around labeling and approval (see 21 CFR 201 ), promotional materials and advertisements are regulated differently (under 21 CFR 202.1(1)(2) Read AgencyIQ’s analysis of this guidance here.] Yes, the guidance confirms.
The intended benefit for sponsors of this approach is that products making use of designated platforms would be eligible to receive expedited development benefits from the FDA, including access to early-stage development meetings with regulators. How can sponsors “prove” that these three criteria are met?
In 1972, when regulatory authority for biologic products was transferred from the National Institutes of Health (NIH) to the FDA, the agency extended the two-study framework to biologic regulation as well. The Heckler case helped to refine the definition of “substantial evidence” in two important ways.
See “Reconciliation of Non-GAAP Measures” for a definition of the terms and reconciliation tables.). 1) See “Reconciliation of Non-GAAP Measures” for a definition of terms and applicable reconciliation tables. The Company’s definition of these non-GAAP measures may differ from similarly titled measures used by others.
BY AMANDA CONTI SEP 13, 2023 1:58 PM CDT Quick background on nonprescription drug regulation Nonprescription drugs, also known as over-the-counter (OTC) drugs, are regulated differently than traditional prescription drugs. The FDA will follow these procedures for both agency-initiated operations (e.g.,
2 The study included patients with mCSPC with both low- and high-volume disease, those who were newly diagnosed, and those who had received prior definitive local therapy or prior treatment with up to six cycles of docetaxel for mCSPC. The recruitment period for the study spanned from December 2015 to July 2017. ERLEADA ® received U.S.
sensitivity and specificity) – including colonoscopy procedures and an FDA-approved stool-based test ( Cologuard ) – but comparatively low uptake, given the prevalence of CRC in the population. Analysis & What’s Next Up front, the advisory committee panel meeting touched on several hot topics in diagnostics regulation.
Over the course of the PHE—which spanned more than three years—the FDA used its expanded authority extensively to issue new direct-to-final guidance documents, creating over 80 in total. The FDA also implemented new flexibilities for certain regulated products and processes, typically via enforcement discretion.
In the study, the definition of severe COVID-19 disease included laboratory-confirmed SARS-CoV-2 and one or more of the following: signs consistent with severe systemic illness, admission to an intensive care unit, respiratory failure, shock, organ failure or death, among other factors. COV2-S (SARS-CoV-2 vaccine) FDAApproval History.
Low Level Light therapy (LLLT), is FDAapproved for treating conditions such as chronic joint pain and slow to heal wounds. (3). Carl was taking ibuprofen and opiates to manage the pain, but knew he had to be careful to regulate it because of the kidney and liver problems it could cause. In the USA at an FDAapproved plant.
Patients who did not meet the definition of confirmed uMRD were randomized 1:1 to receive open-label treatment with ibrutinib or continued ibrutinib + venetoclax. IMBRUVICA is the only FDA-approved medicine in WM and cGVHD. BTK signaling is needed by specific cancer cells to multiply and spread.
OASH concluded “there is widespread current experience with medical use of marijuana in the United States” by licensed healthcare providers “operating in accordance with implemented state-authorized programs, where such medical use is recognized by entities that regulate the practice of medicine under these state jurisdictions.”
What We Expect the FDA to do in December 2023 In this ongoing feature, AgencyIQ looks at public data to determine what the FDA is likely to do in the month ahead, including key deadlines, meetings, events, planned regulations, comment periods and more.
Basics of the bill: FDA’s budget is staying generally level The bill would keep the FDA’s appropriations (i.e., the Congressionally funded portion of the FDA’s total budget) level. billion in “definite user fees.” Per the budget table , this is actually a (relatively) small decrease for the FDA.
With the new LDT final rule, FDA works to clarify what emergency response will look like going forward The Covid-19 pandemic highlighted some gaps in existing policy for how tests and diagnostics are regulated and deployed in emergency situations. In the U.S. IVDs are defined at 21 CFR 809.3(a), The rule amends 21 CFR 809.3(a)
The court also ordered a more definite statement with respect to the manufacturing defect claim. That more definite (except it really wasn’t – more on that later) statement arrived in the form of a Second Amended Complaint. Have you heard the definition of insanity as doing the same thing and expecting a different result?
That Complaint alleges various antivax conspiracy theories concerning COVID-19 vaccines, the FDA, emergency use authorizations, and the media that have circulated since these vaccines first became available. The FDA, however, did not and does not share that belief. FDA (8/23/21) press release (emphasis original). & Com.
FDA , 78 F.4th 2023), was the Fifth Circuit’s blatantly politicized attack on the FDA’sregulation of abortion-related drugs. That’s significant because the labels for over 500 drugs already have such information, under a voluntary FDA program. 4th 210 (5th Cir. Gilead Sciences, Inc. 2023 WL 6390598 (N.D.
In our line of work, much of what we do depends on the continuing validity of how the FDAregulates prescription medical products. It prevents plaintiffs in prescription medical product liability litigation from making collateral attacks on in-force FDA decisions. FDA , 727 F. That’s why Buckman Co. 1, 6 (D.D.C.
It has generally been assumed that such plaintiffs, while free to seek an advisory opinion from the FDA, may not collaterally attack FDA decisions in other litigation. FDA , 727 F. 1989) (refusing to “upset the FDA’s scheme for regulating drugs and cosmetics”); Mitchell v. E.g. , Estee Lauder, Inc. 1, 6 (D.D.C.
The central issue in the Vepuri case was the portion of the conspiracy charge alleging that the three defendants conspired to violate provisions of the Food, Drug and Cosmetic Act that prohibit introducing a “new drug” into interstate commerce unless an FDAapproval is “effective with respect to such drug.” section 355(a).
But the usual is more common than the unusual by definition. When you hear Class III medical device product liability case, you should look for all claims to be dismissed unless there is something as unusual as a basis to claiming the plaintiff’s particular device deviated from its FDA-approved specifications. Stryker Corp. ,
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.
















































Let's personalize your content