This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Food and Drug Administration (FDA) has approved an expanded label for KEYTRUDA, Merck’s anti-PD-1 therapy, as monotherapy for the treatment of patients with locally advanced cutaneous squamous cell carcinoma (cSCC) that is not curable by surgery or radiation. 1) as determined by an FDA-approved test.
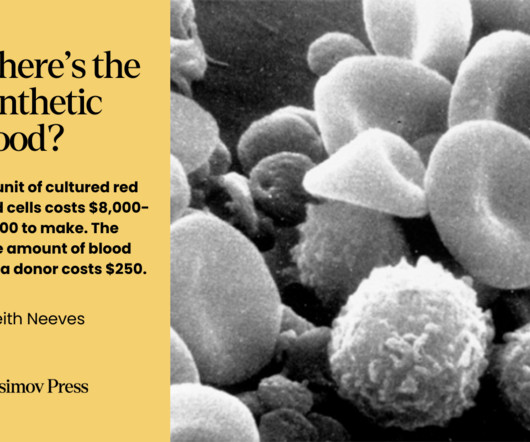
While blood doesn’t fit neatly into the various definitions of an organ , it still comprises a discrete system with specific functions. From Early Transfusion to Component Therapy The earliest documented attempts at blood transfusions involved the transfer of blood from animals to humans in the 17 th century. Always free.
KEYTRUDA is an anti-PD-1 therapy that works by increasing the ability of the body’s immune system to help detect and fight tumor cells. 1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is stage III where patients are not candidates for surgical resection or definitive chemoradiation, or metastatic.
Food and Drug Administration (FDA)-approved test, with disease progression on or after two or more prior lines of therapy including fluoropyrimidine- and platinum-containing chemotherapy and if appropriate, human epidermal growth factor receptor 2 (HER2)/neu-targeted therapy. 1) as determined by an FDA-approved test.
First IL-5 therapyapproved as an add-on treatment in the US for adults with chronic rhinosinusitis with nasal polyps to target eosinophilic inflammation Fourth indication for mepolizumab in the US for eosinophil-driven diseases. The mechanism of action for mepolizumab has not been definitively established.
Adds Rezurock™ (belumosudil) an FDA-approved, first-in-class treatment for adult and pediatric patients 12 years and older with chronic graft-versus-host disease (cGVHD) after failure of at least two prior lines of systemic therapy. Sanofi has entered into a definitive merger agreement with Kadmon Holdings, Inc.
Food and Drug Administration (FDA)-approvedtherapies for treating PAH were primarily vasodilators, designed to overcome the imbalance between vasoactive and vasodilator mediators and to restore endothelial cell function. Historically, the available drugs and U.S. None offers a cure for PAH.
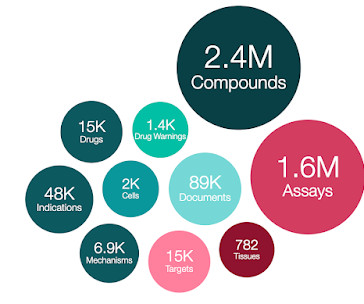
The data in MOLECULE_DICTIONARY.MAX_PHASE now includes consideration of: EMA approved drugs (max_phase=4 for human drugs), USAN clinical candidate drugs (assigned as max_phase = 1 based on USAN guidance that states “Firms usually apply for a USAN when the investigational therapy is in Phase I or Phase II trials”.
NASDAQ: REGN) today announced that the antibody cocktail casirivimab and imdevimab administered together (formerly known as REGN-COV2 or REGEN-COV2), a therapy currently being investigated for use in COVID-19 , has received Emergency Use Authorization (EUA) from the U.S. Food and Drug Administration (FDA). TARRYTOWN, N.Y.,
A rare, or orphan, disease by definition affects a small percentage of the population — fewer than 200,000 people in the U.S. Food and Drug Administration (FDA) approval. Since then, the FDA has significantly changed its approach to rare and orphan diseases. The FDA Since 1983. A Lasting Impact.
Because esophageal cancer generally has poor survival rates, new first-line therapies are urgently needed for these patients,” said Dr. Peter Enzinger, Director, Center for Esophageal and Gastric Cancer, Dana-Farber/Brigham and Women’s Cancer Center. For more information, see “Selected Important Safety Information” below.
The trial investigated neoadjuvant KEYTRUDA, Merck’s anti-PD-1 therapy, plus chemotherapy followed by adjuvant KEYTRUDA as monotherapy (the KEYTRUDA regimen) compared with neoadjuvant chemotherapy followed by adjuvant placebo (the chemotherapy-placebo regimen) in patients with high-risk early-stage triple-negative breast cancer (TNBC).
regulatory landscape for probiotics and other microbiome-based therapies Probiotics, a type of microbiome-based therapy containing live micro-organisms, are broadly available on drug store and grocery store shelves, but the regulatory classifications for products containing the same micro-organism can differ dramatically.
But a new drug launch in the time of COVID-19 comes with definite challenges. Bristol Myers Squibb stated in its press release announcing FDAapproval of the company’s treatment for multiple sclerosis that the commercial launch would be delayed due to the COVID-19 situation. Recently in the U.S.
It is not known whether CD24 on cancer cells has a unique epitope that can be specifically targeted for cancer therapy. By definition, the do-not-eat-me signal refers to phagocytosis of tumour cells by macrophages, although we and others have shown that the CD24/Siglec-10 pathway also regulates the function of T cells and NK cells.
Kymriah was preliminarily granted orphan medicinal product designation by the European Commission (EC) forFL.However, Kymriah would have the occasion to present an important treatment option for those cases with r/ r FL in need of potentially definitive issues, If approved in this implicit third suggestion.
The European Commission (EC) has approved Sanofi and Regeneron’s PD-1 inhibitor Libtayo ® (cemiplimab) for the first-line treatment of adults with non-small cell lung cancer (NSCLC) whose tumor cells have ?50% Patients must have metastatic NSCLC or locally advanced NSCLC and not be a candidate for definitive chemoradiation.
limited efficacy of experimental therapy) Change to the research resulting in increased burden or discomfort New alternative therapy availability (e.g., FDAapproval of a new drug for the condition under study) Impact of participation on alternative therapies (e.g.,
Food and Drug Administration (FDA) approved the use of Aurinia Pharmaceuticals’ Lupkynis TM (voclosporin) as the first oral treatment developed specifically for adults with active lupus nephritis (LN) in combination with standard of care.
Lupus Research Alliance President and CEO Kenneth M.
Radius” or the “Company”) (Nasdaq: RDUS) today announced a definitive agreement to acquire the global development and commercialization rights to Benuvia Therapeutics Inc. ’s PWS is an orphan disease with major endocrine and behavioral manifestations and no FDA-approvedtherapies for the treatment of hyperphagia.
The treatment had been granted breakthrough therapy designation, but FDAapproval would ultimately rest on the sponsor’s ability to demonstrate clinically meaningful improvement.
With today’s FDAapproval, and regulatory approvals or temporary authorizations in approximately 50 additional countries around the world, Veklury® (remdesivir) is one of the tools available today. [ii] ii] The ACTT-1 study sets a high bar for clinical evidence that must be considered as new data emerge.
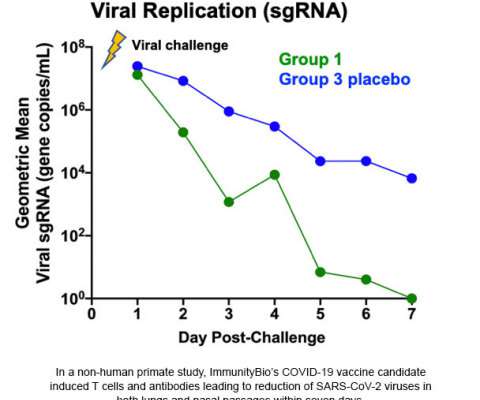
Under the terms of a definitive agreement announced on August 24, 2020, ImmunityBio, Inc. is a late-clinical-stage immunotherapy company developing next-generation therapies that drive immunogenic mechanisms for defeating cancers and infectious diseases. About ImmunityBio and NantKwest Joint Collaboration Agreement. link]
1.
As the legislation explained, the FDA could require the sponsor of an approved “molecularly targeted” medicine to complete a pediatric assessment if the target is “germane” to a pediatric cancer, either in terms of the number of patients or the therapeutic benefit over existing therapies. DORIS MATSUI (D-Calif.)
These follow-on data provided the first definitive prospective evidence demonstrating anti-viral activity for a treatment regimen now available for COVID-19, and also further documented the ability of this treatment to decrease the need for further medical attention,” said George D. who require oxygen therapy due to COVID-19, OR.
. “Patients in our antibody cocktail outpatient clinical trial experienced significant reductions in virus levels and required fewer medical visits for COVID-19, suggesting the therapy can help reduce the current burden on hospitals and healthcare systems,” said George D. who require oxygen therapy due to COVID-19, OR.
First antibody therapy to demonstrate anti-viral effect in patients hospitalized with COVID-19. Patients were randomized to receive the antibody cocktail (either 8,000 mg high dose or 2,400 mg low dose) or placebo, in addition to standard-of-care therapies, with 67% receiving remdesivir and 74% receiving systemic corticosteroids.
Changes in net sales are expressed at constant exchange rates (CER) unless otherwise indicated (definition in Appendix 7) (1) In order to facilitate an understanding of operational performance, Sanofi comments on the business net income statement. Business net income is a non-GAAP financial measure (definition in Appendix 7).
In addition, health-related quality of life (HRQoL), per total Functional Assessment of Cancer Therapy–Prostate (FACT-P), continued to be maintained with ERLEADA ®. Food and Drug Administration (FDA) approval for nmCRPC on February 14, 2018 and was approved for mCSPC on September 17, 2019. ERLEADA ® received U.S.
In 2020, the FDA granted Orphan Drug Designation and Breakthrough Therapy Designation to Esbriet for UILD. Approximately 1 in 10 people living with ILD cannot be given a definitive diagnosis, even after a thorough investigation, and in these cases, they are categorized as having unclassifiable interstitial lung disease (UILD).
For the past three and a half years, Opdivo monotherapy has been an important option that physicians have relied on to address this need and is currently the most commonly used therapy in the post-sorafenib setting,” said Jonathan Cheng, senior vice president and head of oncology development, Bristol Myers Squibb.
NASDAQ: REGN) today announced that the antibody cocktail casirivimab and imdevimab administered together (also known as REGN-COV2 or REGEN-COV2), a therapy currently being investigated for use in COVID-19, has received Emergency Use Authorization (EUA) from the U.S. Food and Drug Administration (FDA). Regeneron Pharmaceuticals, Inc.
BRILINTA is approved in more than 110 countries for the prevention of atherothrombotic events in adult patients with acute coronary syndrome (ACS) and in more than 70 countries for the secondary prevention of cardiovascular events among patients who are at high-risk and have experienced a heart attack. INDICATIONS.
1%) as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations. Patients with EGFR or ALK genomic tumor aberrations should have disease progression on FDA-approvedtherapy for these aberrations prior to receiving OPDIVO. endocrinopathies and dermatologic reactions) are discussed below.
That’s right, if you’re suffering from pain, then “Red Light Therapy” is about to become your new best friend. But what actually is “Red Light Therapy”? Low Level Light therapy (LLLT), is FDAapproved for treating conditions such as chronic joint pain and slow to heal wounds. (3). But it hasn’t gone mainstream yet.
ImmunityBio and NantKwest Joint Collaboration Agreement
Under the terms of a definitive agreement announced on August 24, 2020, ImmunityBio, Inc. is a late-clinical-stage immunotherapy company developing next-generation therapies that drive immunogenic mechanisms for defeating cancers and infectious diseases.
BY AMANDA CONTI | OCT 4, 2023 10:40 PM CDT A quick note: How AgencyIQ gathers data on drug approvals AgencyIQ reviews drug approvals from the Center for Drug Evaluation and Research (CDER) and the Center for Biologics Evaluation and Research (CBER). The FDA also refers to novel products as “New Molecular Entities,” or NMEs.
AbbVie also presented data from the Phase 3 U-ACHIEVE and U-ACCOMPLISH studies evaluating the efficacy and safety of Rinvoq (45 mg, once daily) as induction therapy in patients with moderate to severe UC, which highlighted the impact of Rinvoq on clinical, endoscopic and histologic outcomes after 8 weeks of treatment.
Heckler in 1986, several manufacturers sued the FDA after it ordered them to remove several indications from the labels of their oral proteolytic enzymes (OPE) products for lack of evidence. The Heckler case helped to refine the definition of “substantial evidence” in two important ways.
For this reason, the guidance advises sponsors that “some technologies that industry and FDA have historically considered to be platform technologies might not meet the statutory definition and statutory eligibility factors and, if not, would not be eligible for the designation program.”
“Combining our novel therapies to deliver a new potential treatment is an example of AbbVie’s innovative approach to identify options for difficult-to-treat blood cancers, like CLL,” said Mohamed Zaki , M.D., IMBRUVICA is the only FDA-approved medicine in WM and cGVHD.
In a first, FDA tackles treatments for stimulant use disorder A new draft guidance from the FDA gives developers a roadmap to advance the development of novel therapies to address stimulant use disorders. While stimulant use disorder is increasing, there are currently no FDA-approved medications.
Today’s data, involving an additional 524 patients from the ongoing Phase 2/3 trial, provides definitive final virology results and meets the clinical endpoint of reducing medical visits. TARRYTOWN, N.Y. , 28, 2020 /PRNewswire/ — . Regeneron has shared these results with the U.S. combined dose groups; 6.5% placebo; p=0.024).
Until recently, the FDA relied on a monograph process through which firms could bring OTC drugs to market without FDAapproval so long as it adhered to pre-set terms under the monograph. CHPA considered the original studies to be sound and instead found issues with the newer studies.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.
















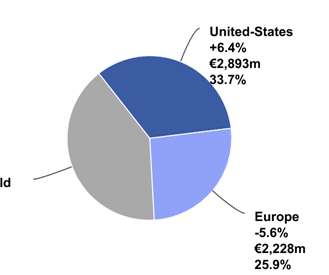




















Let's personalize your content