This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Food and Drug Administration (FDA)-approved therapies for treating PAH were primarily vasodilators, designed to overcome the imbalance between vasoactive and vasodilator mediators and to restore endothelial cell function. Their early definition — as well as plans for recording and tracking — is a major factor in a trial’s success.
Food and Drug Administration (FDA) has approved an expanded label for KEYTRUDA, Merck’s anti-PD-1 therapy, as monotherapy for the treatment of patients with locally advanced cutaneous squamous cell carcinoma (cSCC) that is not curable by surgery or radiation. 1) as determined by an FDA-approved test.
PROs in clinical trials are important as they capture the patient’s perspective and ensure that the impact of an intervention is comprehensively evaluated. Food and Drug Administration (FDA) increasingly look to patients to understand how they describe their health status. What are PROs in clinical trials?
The data in MOLECULE_DICTIONARY.MAX_PHASE now includes consideration of: EMA approved drugs (max_phase=4 for human drugs), USAN clinical candidate drugs (assigned as max_phase = 1 based on USAN guidance that states “Firms usually apply for a USAN when the investigational therapy is in Phase I or Phase II trials”.
While blood doesn’t fit neatly into the various definitions of an organ , it still comprises a discrete system with specific functions. However, the subsequent death of another patient thrust Denys into a contentious trial. The results of the trial are expected at the end of 2024. Data from Rousseau G.F.
Additionally, mepolizumab was the first biologic therapy indicated for adults with eosinophilic granulomatosis with polyangiitis (EGPA) and also the first biologic to be approved for patients aged 12 years and older with hypereosinophilic syndrome (HES). With 41 clinical trials, mepolizumab has been studied in over 4,000 patients.
There are currently more than 1,400 trials studying KEYTRUDA across a wide variety of cancers and treatment settings. 1%] as determined by an FDA-approved test, with no EGFR or ALK genomic tumor aberrations, and is stage III where patients are not candidates for surgical resection or definitive chemoradiation, or metastatic.
accelerated approval indication for KEYTRUDA for the treatment of patients with recurrent locally advanced or metastatic gastric or gastroesophageal junction (GEJ) adenocarcinoma whose tumors express PD-L1 [combined positive score (CPS ?1)] 1)] as determined by a U.S. Merck has the industry’s largest immuno-oncology clinical research program.
1 Sairiyo has an exclusive license from a research and development organization to develop and commercialize reformulated Cepharanthine for all diseases and exclusive rights to the patent, method of manufacturing, clinical supply, pre-clinical data and know-how to support FDA clinical trials.
A rare, or orphan, disease by definition affects a small percentage of the population — fewer than 200,000 people in the U.S. Food and Drug Administration (FDA) approval. Since then, the FDA has significantly changed its approach to rare and orphan diseases. The FDA Since 1983.
Approval based on a Phase 3 trial demonstrating Libtayo significantly improved overall survival compared to chemotherapy in advanced NSCLC that included challenging-to-treat patient populations Libtayo now approved by the European Commission for three advanced cancers. 50% PD-L1 expression and no EGFR, ALK or ROS1 aberrations.
As in earlier outpatient trial, immune status when patients entered the trial was a strong predictor of viral load and clinical outcomes. The primary clinical objective of this initial analysis was to determine if there was sufficient efficacy in these patients to warrant continuing the trial (i.e., futility analysis).
The approval is based on results from the Phase 3 KEYNOTE-590 trial, which demonstrated significant improvements in overall survival (OS), progression-free survival (PFS) and objective response rate (ORR) for KEYTRUDA plus fluorouracil (FU) and cisplatin versus FU and cisplatin alone, regardless of histology or PD-L1 expression status.
Kymriah was preliminarily granted orphan medicinal product designation by the European Commission (EC) forFL.However, Kymriah would have the occasion to present an important treatment option for those cases with r/ r FL in need of potentially definitive issues, If approved in this implicit third suggestion.
With today’s FDAapproval, and regulatory approvals or temporary authorizations in approximately 50 additional countries around the world, Veklury® (remdesivir) is one of the tools available today. This type of study reduces the potential for bias and provides the highest quality scientific evidence.
Adds Rezurock™ (belumosudil) an FDA-approved, first-in-class treatment for adult and pediatric patients 12 years and older with chronic graft-versus-host disease (cGVHD) after failure of at least two prior lines of systemic therapy. Sanofi has entered into a definitive merger agreement with Kadmon Holdings, Inc.
The trial investigated neoadjuvant KEYTRUDA, Merck’s anti-PD-1 therapy, plus chemotherapy followed by adjuvant KEYTRUDA as monotherapy (the KEYTRUDA regimen) compared with neoadjuvant chemotherapy followed by adjuvant placebo (the chemotherapy-placebo regimen) in patients with high-risk early-stage triple-negative breast cancer (TNBC).
Clinical trials for ultra-rare diseases can be particularly challenging to mount due to small, geographically-dispersed patient populations. For such trials, the US Food and Drug Administration (FDA) may allow the use of credible real-world data (RWD) and real-world evidence (RWE) in lieu of data collected in a Phase 3 trial.
RAD011 is a pivotal-trial ready synthetic cannabidiol oral solution with potential utilization in multiple endocrine and metabolic orphan diseases.
Prader-Willi syndrome (“PWS”) will be the initial indication, which has been granted Orphan Drug and Fast Track Designation by the FDA.
BOSTON, Jan.
This year we completed investigational new drug (IND) enabling studies of ONC-841, which is an antagonist of Siglec-10, and received US Food and Drug Administration (FDA) approval for first-in-human study (FIH) in patients with solid tumours. Biomarkers are the holy grail of clinical trials.
Food and Drug Administration (FDA) approved the use of Aurinia Pharmaceuticals’ Lupkynis TM (voclosporin) as the first oral treatment developed specifically for adults with active lupus nephritis (LN) in combination with standard of care.
NEW YORK , Jan.
Lupus Research Alliance President and CEO Kenneth M.
Today’s data, involving an additional 524 patients from the ongoing Phase 2/3 trial, provides definitive final virology results and meets the clinical endpoint of reducing medical visits. REGN-COV2 was well tolerated in the trial. TARRYTOWN, N.Y. , 28, 2020 /PRNewswire/ — . Regeneron Pharmaceuticals, Inc.
The sponsor is the pharmaceutical company conducting the trial. If you mean using a different contract research organization (CRO) for the different phases of clinical trials – that’s different. Also consider CRO oversight, trial management, data handling and record keeping, as well as allocation of responsibilities.
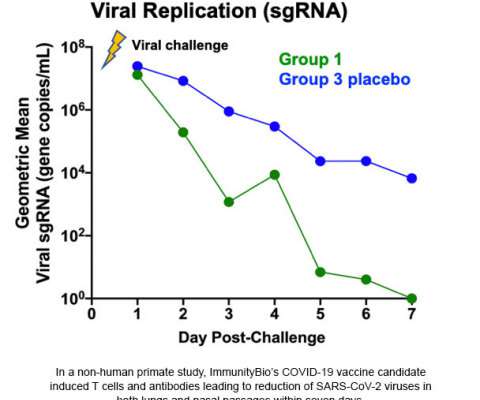
Pending discussions with the FDA, the oral vaccine will enter Phase I trials as a prime and boost, and will be explored to provide a boost to subcutaneous vaccinations. For more information about ImmunityBio’s COVID-19 vaccine trials, please contact clinicalresearch@hoag.org. and its affiliate NantKwest, Inc.
Food and Drug Administration (FDA) to begin a Phase I clinical trial of hAd5-COVID-19, the company’s novel COVID-19 vaccine candidate that targets both the inner nucleocapsid (N), engineered to activate T cells, and outer spike (S) protein, engineered to activate antibodies against the coronavirus (SARS-CoV-2). i,ii,iii,iv.
January 29, 2021 – Johnson & Johnson (NYSE: JNJ) (the Company) today announced topline efficacy and safety data from the Phase 3 ENSEMBLE clinical trial, demonstrating that the investigational single-dose COVID-19 vaccine in development at its Janssen Pharmaceutical Companies met all primary and key secondary endpoints.
NASDAQ: REGN ) today announced that the New England Journal of Medicine (NEJM) has published initial clinical data from an ongoing seamless Phase 1/2/3 trial of the antibody cocktail casirivimab and imdevimab in non-hospitalized patients with COVID-19. TARRYTOWN, N.Y. , 17, 2020 /PRNewswire/ — Regeneron Pharmaceuticals, Inc.
The clinical evidence from Regeneron’s outpatient trial suggests that monoclonal antibodies such as casirivimab and imdevimab have the greatest benefit when given early after diagnosis and in patients who have not yet mounted their own immune response or who have high viral load.
If a drug was previously approved for cancer, but is now approved for a cardiology condition, it would not be considered novel. The FDA also refers to novel products as “New Molecular Entities,” or NMEs. Data on these novel approvals is published throughout the year by both CDER and CBER. of all NME approvals this year.
Remdesivir gets full FDAapproval to treat COVID. Remdesivir’s full approval Thursday by the FDA comes after the agency granted it emergency use authorization last spring. National Institutes of Health, the FDA announced in a statement. It is given intravenously to hospitalized patients.
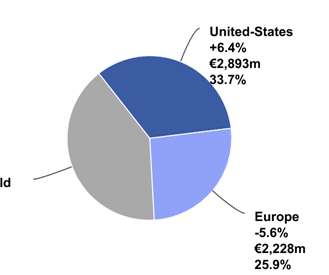
Changes in net sales are expressed at constant exchange rates (CER) unless otherwise indicated (definition in Appendix 7) (1) In order to facilitate an understanding of operational performance, Sanofi comments on the business net income statement. Business net income is a non-GAAP financial measure (definition in Appendix 7).
So what about all of the “biotic” terms that pop up in the news, on products labels and even in FDA warning letters? Unfortunately, there is no regulatory body that governs the terminology used in this space, although there are definitions that are generally accepted by the scientific community.
Remdesivir gets full FDAapproval to treat COVID-19. Remdesivir’s full approval Thursday by the U.S. National Institutes of Health, the FDA announced in a statement. CDC widens definition of ‘close contact’ for tracking COVID-19 infections. 22 news release.
Traditionally, FDA has interpreted the need for “well-controlled investigations” to mean at least two clinical trials for new drugs, or applications for supplemental indications. The Heckler case helped to refine the definition of “substantial evidence” in two important ways.
“Patients in our antibody cocktail outpatient clinical trial experienced significant reductions in virus levels and required fewer medical visits for COVID-19, suggesting the therapy can help reduce the current burden on hospitals and healthcare systems,” said George D. .” Definition of High-Risk Patients.
In 2017, Opdivo was granted accelerated approval by the FDA as a single agent for patients with HCC who have been previously treated with sorafenib. The accelerated approval was based on tumor responses from the Phase 1/2 CheckMate -040 trial.
“We are working closely with the FDA in hopes of offering Esbriet to people with UILD, a rare and debilitating disease.”. The sNDA is based on results from a pivotal, 24-week Phase II trial, which was the first randomized controlled study specifically designed and conducted solely in people with UILD. About the Pivotal Study.
Opdivo was first granted this indication in 2017 under the FDA’s accelerated approval program, making it the first immunotherapy agent to be approved for use in this setting. The accelerated approval was based on tumor responses from the Phase 1/2 CheckMate -040 trial.
FDA’s guidance on developing products to prevent or treat Covid-19 Of the five guidance documents that received an extension, one addresses the development of drugs and biological products for Covid-19. Lastly, the revised guidance briefly discussed trials meant to assess drugs intended for prevention of Covid-19.
Dietary supplements are intended for ingestion only, meaning that products that are administered through other routes, such as topically, by injection or by inhalation, do not fall within this definition. L-glutamine offers an example of a more recently approved drug product.
AbbVie also presented an updated analysis from the Phase 3 ARTEMIS studies assessing the efficacy and duration of Durysta (bimatoprost intracameral implant), the first and only FDA-approved dissolvable implant to reduce eye pressure in people with open angle glaucoma or high eye pressure. years in patients with RA.
The approval by the US Food and Drug Administration (FDA) was based on positive results from the THALES Phase III trial that showed aspirin plus BRILINTA 90mg significantly reduced the rate of the composite of stroke and death compared to aspirin alone in patients with acute ischemic stroke or TIA. for aspirin alone.
The clinical evidence from Regeneron’s outpatient trial suggests that monoclonal antibodies such as REGEN-COV2 have the greatest benefit when given early after diagnosis and in patients who have not yet mounted their own immune response or who have high viral load. Data from these trials will be used to support a future BLA submission.
While stimulant use disorder is increasing, there are currently no FDA-approved medications. The meeting discussed the challenges associated with running trials for substance use disorder medications, from medication nonadherence to trial enrollment.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.



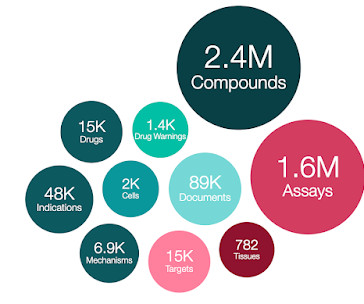
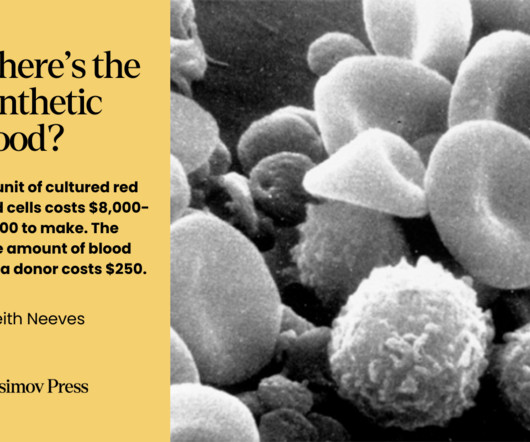



































Let's personalize your content