This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Pharmacies and Pharmacy Benefit Managers is a definitive, nonpartisan resource that includes the most current information about pharmacy dispensing channels, third-party payers, pharmacy benefit managers (PBMs), patients’ financial contributions, government regulations, and much more. The 2022 Economic Report on U.S. Click to Enlarge].
Pharmacies and Pharmacy Benefit Managers is a definitive, nonpartisan resource that includes the most current information about pharmacy dispensing channels, third-party payers, pharmacy benefit managers (PBMs), patients’ financial contributions, government regulations, and much more. The 2023 Economic Report on U.S.
Lenz, Principal Medical Device Regulation Expert & Sophia R. Gibbs — For more than three decades, FDA has claimed that the Federal Food, Drug & Cosmetic (FD&C Act) gives the agency legal authority to regulate laboratory developed tests (LDTs) as medical devices (see our prior post here ). Gaulkin & Jeffrey N.
regulator lays out proposal for international device and diagnostics recognition The British medical device regulator just issued its promised framework on international recognition. law as the Medical Devices Regulation 2002 (UK MDR 2002). law as the Medical Devices Regulation 2002 (UK MDR 2002).
1 Sairiyo has an exclusive license from a research and development organization to develop and commercialize reformulated Cepharanthine for all diseases and exclusive rights to the patent, method of manufacturing, clinical supply, pre-clinical data and know-how to support FDA clinical trials. PharmaDrug Inc.
Kirschenbaum — Last Friday, May 26, CMS published in the Federal Register an assortment of proposals to change the regulations governing the Medicaid Drug Rebate Program. Most are new or revised definitions and administrative changes, but several proposals represent new policies that should be of concern to drug manufacturers.
Amongst other things, FDA co-opted many of the same definitions for key terms for implementation of the BPCIA. FDA borrowed this definition from 21 C.F.R. FDA replied that its definition of strength including concentration was clear even in 2009. FDA explained that its bioequivalence regulations at 21 C.F.R.
While the Federal Food, Drug & Cosmetic Act does not explicitly define “advertisement,” FDA provides several examples in its regulations at 21 CFR § 201.1(l)(1) We note this clarification is helpful given the modification to the definition of “promotional labeling” in the Revised Draft Guidance as part of footnote 4. l)(1) (e.g.,
The 51 regulations that FDA is currently working on The FDA today unveiled its much-anticipated Spring 2023 Unified Agenda, a document outlining the regulations the agency plans to release in 2023 and beyond. The anticipated date of publication is June 2023, meaning we should see this regulation imminently.
By Riëtte van Laack — The dietary supplement exclusionary clause is, as its name suggests, a clause in the Federal Food, Drug, and Cosmetic Act (FDC Act) definition of dietary supplement. That clause excludes those ingredients that were first marketed as drug ingredients. What led to NPA’s lawsuit? The facts are detailed in the complaint.
Before I begin, I just want to caveat everything with the fact that HIPAA is a complex regulation open to interpretation, and in the end your legal and compliance teams need to be comfortable with how you handle data and the risk associated with those methods. The other part of the definition is what constitutes “health information.”
Determining if you have a combination product The high-level definition of combination product seems straightforward: a product with at least two constituent parts, such as: Drug/biologic (e.g., metered-dose inhaler) However, the four-part definition in 21 CFR 3.2(e) therapeutic drug/monoclonal antibody) Drug/device (e.g.,
government was put in place on February 27, 2023 and includes a number of proposed regulations to address trade relations between the E.U. First: all medicines for Northern Ireland must be approved by the MHRA The new regulation (EU) 2023/1182 applies to medicines that require marketing authorization. Definitions from the existing E.U.
Regulators wanted input on questions related to data access and re-use, to inform its legislative framework on common European data spaces. Two intertwined proposals have been laid out – a regulation on data governance and the proposed data act. Important definitions come both within the proposed law and from other Union legislation.
The standard for the audit used depends on the type of vendor and would include the applicable regulations and ICH guidelines. Q: What is the definition of a drug? The term drug substance and drug product are found in the NDA regulations 21 CFR 314.3, A: For FDA 1572 is a Statement of Investigator under US Regulations/Laws.
BY LAURA DIANGELO, MPH | JUL 10, 2024 3:47 PM CDT Regulatory background: The FDA is responsible for overseeing information about regulated products. The FDA is responsible for ensuring that medical products are adequately labeled in accordance with federal regulations, including the product’s “intended use” and relevant safety information.
Alexion ) have entered into a definitive agreement for AstraZeneca to acquire Alexion. Neither this announcement nor any copy of it may be taken or transmitted directly or indirectly into or from any jurisdiction where to do so would constitute a violation of the relevant laws or regulations of such jurisdiction.
For context, while prescription drug products are required to comply with FDA’s regulatory requirements around labeling and approval (see 21 CFR 201 ), promotional materials and advertisements are regulated differently (under 21 CFR 202.1(1)(2) Read AgencyIQ’s analysis of this guidance here.]
within the CSA’s definition of “marihuana” or “marijuana” based on FDA’s eight factor analysis. Marijuana” outside of the CSA definition, including hemp, mature stalks, fiber from the mature stalks, oil or cake made from seeds, and sterilized seeds incapable of germination are not controlled and not subject to the recommendation.
MHRA selects eight technologies to help it test its new innovative devices pathway The British regulator and its partners just released the list of products and companies chosen to participate in the pilot of its Innovative Device Access Pathway (IDAP). device regulations and the pilot for innovative products. and abroad.
The Final Guidance clarifies that, although both applicable drugs and selected drugs must be covered under a Discount Program agreement, selected drugs are excluded from the definition of an applicable drug, so they are not subject to applicable discounts during an MFP applicability year. Non-applicable drugs (e.g.,
Compliance and Regulation: Azure AD B2C provides compliance with industry regulations such as GDPR and HIPAA, as well as support for authentication standards like OpenID Connect and OAuth 2.0. This makes it easier for businesses to comply with regulatory requirements and ensure the security of user data.
The intended benefit for sponsors of this approach is that products making use of designated platforms would be eligible to receive expedited development benefits from the FDA, including access to early-stage development meetings with regulators. based on a letter of authorization from the application holder).”
AgencyIQ provides a status update for regulated industry. Intro: The FDA’s rule on diagnostics and what it means for developers A quick intro about how FDA regulates In vitro diagnostics (IVDs) and Laboratory Developed Tests (LDTs): In the U.S., diagnostic products are regulated as medical devices. The rule amends 21 CFR 809.3(a)
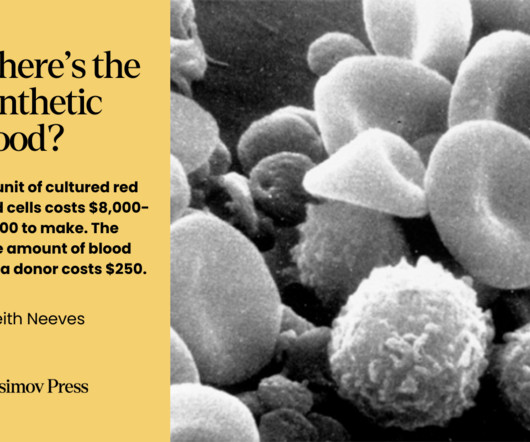
While blood doesn’t fit neatly into the various definitions of an organ , it still comprises a discrete system with specific functions. It is licensed in Russia and South Africa and is included in a few compassionate use programs across the globe, meaning it can be used clinically in cases where patients have few remaining options.
FDA responses to such requests represent the FDA’s best judgment about how a device would be regulated, based upon review of information provided by a requester, including the description of the device and its intended use. Should the FDA grant the De Novo classification request, the Class II device would be cleared to be marketed.
Unfortunately, there is no regulatory body that governs the terminology used in this space, although there are definitions that are generally accepted by the scientific community. Postbiotic: “Preparation of inanimate micro-organisms and/or their components that confers a health benefit on the host,” according to the ISAPP definition.
The FDA regulates both product labeling ( 21 CFR 201 ) and promotional labeling ( 21 CFR 202 ). FDA anticipated at the time that most PDURS output would be promotional labeling, and therefore “would only be required to be submitted at the time of initial dissemination, pursuant to these existing regulations.”
Radius” or the “Company”) (Nasdaq: RDUS) today announced a definitive agreement to acquire the global development and commercialization rights to Benuvia Therapeutics Inc. ’s Within PWS, cannabidiol targets signaling pathways and receptors that regulate the physical symptoms of hyperphagia and anxiety.
BOSTON, Jan.
For example, in September 2022 the FDA finalized a guidance document on how sponsors of drug product submissions could effectively flag RWE for regulators. The group also called on the FDA to “provide examples of studies and how they may be treated as per the guidance and whether the IND regulations apply.”
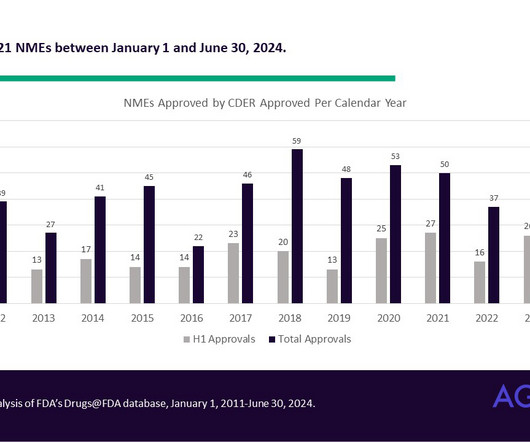
All products are received in one of two forms: A New Drug Application (or NDA, for pharmaceuticals) or a Biologics License Application (BLA, for biologics). While the definition of NME has changed over the years, it can sometimes include a combination product consisting of at least one drug that has previously been approved.
On July 27, Pfizer and BioNTech announced that following extensive review of preclinical and clinical data from Phase 1/2 clinical trials, and in consultation with the FDA’s Center for Biologics Evaluation and Research (CBER) and other global regulators, the companies selected the BNT162b2 vaccine candidate to move forward into a Phase 2/3 study.
BY AMANDA CONTI | JAN 10, 2024 9:29 PM CST Quick background: Goal dates under the Prescription Drug User Fee Act (PDUFA) The FDA collects user fees as part of an essential bargain between regulators and industry. According to Downey, the timeline of pre-license inspections for biologics can create challenges.
See “Reconciliation of Non-GAAP Measures” for a definition of the terms and reconciliation tables.).
Pipeline progress is expected across the vaccines, therapeutics, and devices portfolios, anticipating at least one Phase 3 launch and one Biologics License Application (BLA)/Emergency Use Authorization (EUA) filing.
The regulator sent the rule to the White House’s Office of Information and Regulatory Affairs (OIRA) on October 4, 2022. The content of the PMI : The regulation describes, in broad terms, what must be included in the PMI. However, only licensed blood establishments would be required to submit PMI to FDA for approval.”
Companies are paying for performance here, and it’s reasonable for them to expect performance” remarked Burgess, “And then we hear other discussions that the FDA wants to vastly expand its authority into licensing laboratory developed tests. If you don’t have the staff to do this, how are you going to have the staff to do that?”
The Executive Director of the Institute for Behavior Change (IBC), licensed psychologist and certified school psychologist Steve Kossor, has been involved in the planning and delivery of what became known as Intensive Behavioral Health Services (IBHS) in Pennsylvania since 1981. push(arguments)},i[r].l=1*new Want to work with us?
Sponsors would , if selected for the pilot, “receive more frequent advice related to such specific issues through additional interactions to facilitate novel drug and biological product program development and generate high quality and reliable data intended to support a Biologics License Application (BLA) or New Drug Application (NDA).”
Background and reason
Moberg Pharma is a Specialty Pharma company focused on developing and commercialising proprietary, acquired and licensed products globally, from clinical development of products based on proven substances to commercialisation. CET on November 6, 2020. This information was brought to you by Cision [link].
Amgen develops product candidates internally and through licensing collaborations, partnerships and joint ventures. Furthermore, Amgen’s research, testing, pricing, marketing and other operations are subject to extensive regulation by domestic and foreign government regulatory authorities. J Intern Med 2012;272:121–32.
Amgen develops product candidates internally and through licensing collaborations, partnerships and joint ventures. Furthermore, Amgen’s research, testing, pricing, marketing and other operations are subject to extensive regulation by domestic and foreign government regulatory authorities. 2019;12(5):539-549.
Eur Respir J.
OASH concluded “there is widespread current experience with medical use of marijuana in the United States” by licensed healthcare providers “operating in accordance with implemented state-authorized programs, where such medical use is recognized by entities that regulate the practice of medicine under these state jurisdictions.”
What We Expect the FDA to do in December 2023 In this ongoing feature, AgencyIQ looks at public data to determine what the FDA is likely to do in the month ahead, including key deadlines, meetings, events, planned regulations, comment periods and more. The following PDUFA dates were obtained from publicly available sources.
Most research to date has provided doses in a highly controlled, positive environment, often with a licensed mental health practitioner present to help guide the participant through the experience. The April 2024 multi-day workshop showcased presentations and panels from various regulators, trade groups, academics and patient advocates.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.













































Let's personalize your content