This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
By Riëtte van Laack — FDA regulates pet food similar to other animal foods. As anyone familiar with pet (and other animal) food regulation knows, many states require premarket label review and approval and registration of the manufacturer/distributor and/or product for a fee.
By Véronique Li, Senior Medical Device Regulation Expert & Ana Loloei & Allyson B. Mullen — More than five years after FDA first announced its plan to harmonize 21 CFR Part 820 with ISO 13485, on February 2, 2024, FDA finally issued the Quality Management System Regulation (QMSR) Final Rule. The new § 820.10 Revised § 820.3
While there is no universally accepted definition for rare disease, it is estimated that more than 10,000 distinct rare conditions exist, and people living with these rare diseases represent as much as 10% of the global population. In rare diseases, evidence gaps are often significant, necessitating de novo evidence generation.
Commission unveils “one substance, one assessment” reform package The Commission has long aspired to realize its one substance, one assessment concept in the EU’s chemical regulatory regime. With the publication of three proposed legal acts, this concept is one step closer to streamlining the way the bloc regulates chemicals.
California publishes draft regulations for landmark plastic pollution reduction act CalRecycle has launched the formal rulemaking process for Senate Bill 54, which will implement a sweeping plan to reduce plastic waste in the state by 2032. By 2028, the state will require the recycling of 30% of single-use packaging and food service ware.
The FDA has determined that ophthalmic dispensers are now regulated as devices, and the drug and device together are regulated as a combination product. Therefore, the FDA will examine product classifications, especially for those products that have been regulated as drugs even though they may satisfy the device definition.
PFAS regulation in California (late 2023 edition) California consistently maintains its status as both one of the most important economies in the world as well as one of the most regulated states in the United States. In 2019, the state began to significantly ramp up its PFAS regulation and research.
Manufactured since the 1940s, their carbon-fluorine bond tends to be the hallmark of a PFAS (though there is no common definition). As such, they can be found in myriad applications ranging from raincoats, refrigerants, and lubricants to non-stick pans, food packaging, and semiconductors. PFAS regulation in the E.U.
New guidance provides definition for orphan device, offers alternative trial designs New guidance from the European Commission outlines alternatives for full pre-market clinical trials for orphan devices, defined by the Commission for the first time. Does this guidance offer the formal definition of “orphan device” we’ve been waiting for?
In a previously-published Phase 2 study evaluating patients with UC, mirikizumab down-regulated several gene transcripts associated with inflamed mucosa and up-regulated gene transcripts correlated with healthy mucosa and markers of functional healing after 12 weeks, as defined by clinical disease indices of endoscopy and histology.
Jan Schakwosky has introduced the No Toxics in Food Packaging Act of 2023, which seeks to ban the use of ortho-phthalates, per- and polyfluoroalkyl substances, bisphenol compounds, styrene, and antimony trioxide as food contact substances. No Toxics in Food Packaging Act of 2023 On October 26, 2023, Reps.
Determining if you have a combination product The high-level definition of combination product seems straightforward: a product with at least two constituent parts, such as: Drug/biologic (e.g., metered-dose inhaler) However, the four-part definition in 21 CFR 3.2(e) The drug product is packaged and shipped as a solid powder.
Gaulkin — In May 2023, we posted about a CMS proposed regulation that sought to make a wide variety of changes to the Medicaid Drug Rebate Program (MDRP), including a new “price verification survey,” and a controversial proposal to require “stacking” of discounts to different customers when determining best price.
Cannabis groups to Congress: FDA should regulate CBD as a dietary supplement U.S. legislators have asked for help to reimagine how the FDA should regulate cannabidiol (CBD) following the agency’s determination that it could not make use of its existing legislative or regulatory authorities to do so. percent on a dry weight basis. ”
In this second of our two-part series, we continue our discussion about significant recent developments regarding the regulation of ophthalmic products and discuss what to expect in the months ahead. All regulated parties, including ophthalmic drug and device manufacturers, should take note. Unsurprisingly, the U.S. injectables).
The 51 regulations that FDA is currently working on The FDA today unveiled its much-anticipated Spring 2023 Unified Agenda, a document outlining the regulations the agency plans to release in 2023 and beyond. The anticipated date of publication is June 2023, meaning we should see this regulation imminently.
The standard for the audit used depends on the type of vendor and would include the applicable regulations and ICH guidelines. Q: What is the typical time period between the submission of the briefing package and the pre-IND meeting? Q: What is the definition of a drug? Yes, each drug has its own requirements.
An AgencyIQ explainer on the EPA’s definition of PFAS may be reviewed here.] Background: PFAS regulation in California PFAS have been the subject of significant regulations in California, beginning in the 2010s [for an in-depth review of PFAS regulation in California, see the AgencyIQ analysis here ].
In a new final rule, FDA carves out a regulatory niche for medical gases Industry has been lobbying FDA and Congress to regulate medical gases different from other types of drug products since the 1970s. It also proposed several packaging changes to safeguard against misidentification. The Medical Gas Safety Act , introduced by Rep.
government was put in place on February 27, 2023 and includes a number of proposed regulations to address trade relations between the E.U. First: all medicines for Northern Ireland must be approved by the MHRA The new regulation (EU) 2023/1182 applies to medicines that require marketing authorization. Definitions from the existing E.U.
During that meeting, the sponsor presented data by endpoint, which complemented the data package and indicated the benefits of treatment outweighed the risks. The agency encouraged further review of the data for potential reference as a historical control. The FDA also granted a second meeting for review.
Generally, how have states attempted to regulate PFAS? States are attempting to regulate PFAS in a variety of ways, including via multiple in-state regulatory and legislative avenues. Used products and used product components are officially exempted from the regulations, as well as manufacturers that employ 25 or fewer people.
As the Pfizer and BioNTech COVID-19 vaccine goes to regulators and the Moderna vaccine approaches the end of its Phase III trials, AstraZeneca and the University of Oxford announced high-level results from an interim analysis of their COVID-19 vaccine, AZD1222. Pascal Soriot, CEO of AstraZeneca, pictured above.
California’s definition of ingredient is the same as defined at the federal level which is, any single chemical entity or mixture used as a component in the manufacture of a cosmetic product ( 21 CFR 700.3(e) A definition of the term “hold” is absent from both of these bills. Chapter 54.
Recently introduced legislation North Carolina: On May 1, 2024, North Carolina State Representative JOHN AUTRY introduced HB 973 , a bill to ban intentionally added PFAS in food packaging. If passed, the law would go into effect in January 2025.
What’s going on in the world of PFAS – early 2024 edition This feature from AgencyIQ focuses on the wide world of per- and polyfluoroalkyl substance regulation, legislation, and litigation around the United States. An AgencyIQ explainer on the EPA’s definition of PFAS may be reviewed here.] since June 21, 2006.
Just over a month ago, the European Commission released its proposal for the new pharmaceutical directive and regulation. BY KIRSTEN MESSMER, PHD, RAC JUN 5, 2023 10:14 PM CDT Regulatory Background: How things work now Regulation (EC) No 141/2000 (the Orphan Regulation) provides the legal framework for orphan drug designations and incentives.
The standards would, as stated previously, be voluntary, unless mandated by statute or regulation, and they cannot conflict with existing law or regulation. Consensus does not require unanimity, but a general agreement. Existing published VCS may be identified internally by FDA or externally by stakeholders.
A 2017 European Parliament resolution called on the European Commission and the European Council “to formulate a better definition of the concept – and analyse the causes – of shortages of medicines.” Following this regulation, the EMA now coordinates shortage prevention, management and communication efforts among Member States.
and internationally Establishing and communicating a substance’s potential to cause cancer is a cornerstone of chemical regulation worldwide. Many entries in category 2B are only weakly associated with carcinogenicity and are generally unlikely to be regulated for causing cancer. For more on the regulation of glyphosate in the E.U.,
Almost 10 years late, the new framework’s arrival will represent significant changes in the regulation of these pesticide substances. Active substances that have been approved for use in the EU are listed in the Annex of Regulation (EU) No 540/2011; currently, approximately 450 entries are listed.
NOV 8, 2023 10:02 PM CST Regulatory background The Federal Food, Drug, and Cosmetics Act (FD&C Act) is administered by the Food and Drug Administration (FDA) and regulates cosmetics. . | BY PATRICIA ISCARO, ESQ. | MoCRA also provides labeling, reporting, and recordkeeping requirements. [
Regulation (EC) No 141/2000 (the Orphan Regulation) provides the legal framework for orphan drug designations and incentives.The Orphan Regulation entered into force in January, 2000, outlining requirements for orphan designation and incentives, and establishing the EMA Committee for Orphan Medicinal Products (COMP). regulators.
The agency has kept the expected publication date for the National Primary Drinking Water Regulation (NPDWR) for six PFAS consistent between the Fall 2022 and Spring 2023 Unified Agendas [for an in-depth analysis on the proposed NPDWR, see this AgencyIQ piece ]. To contact the editor of this analysis, please email Patricia Iscaro.
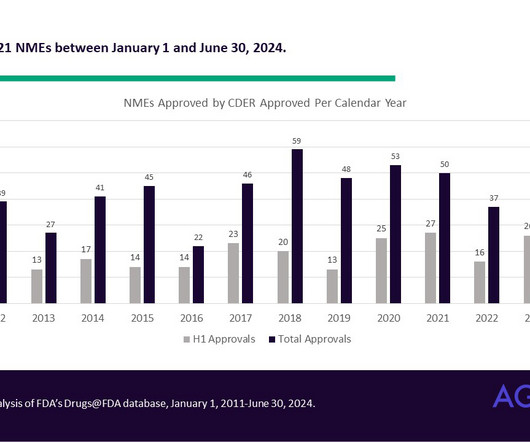
While the definition of NME has changed over the years, it can sometimes include a combination product consisting of at least one drug that has previously been approved. AgencyIQ compiles these data using information in approval letters, labeling and review packages posted to the Drugs@FDA database.
Unfortunately, there is no regulatory body that governs the terminology used in this space, although there are definitions that are generally accepted by the scientific community. Postbiotic: “Preparation of inanimate micro-organisms and/or their components that confers a health benefit on the host,” according to the ISAPP definition.
These are the lists of national and international standards the Commission plans to harmonize with its device and diagnostics regulations. But standards harmonized under the directives may not be used to show compliance with the new Medical Device Regulation, which replaced the MDD. This is similar to the situation in the U.S.,
Within the realm of FDA-required labeling, there are currently a few different types of information a sponsor might develop specifically for patient use: medication guides, instructions for use (IFU), consumer medical information (CMI) and patient package inserts (PPI). The FDA recently concluded its work on a proposed rule focused on PMI.
A 2017 European Parliament resolution called on the European Commission and the European Council “to formulate a better definition of the concept – and analyse the causes – of shortages of medicines.” Regulation (EU) 2022/123 became applicable on March 1, 2022, with the exception of most provisions related to medical devices.
FDA updates set the stage for broader use of harmonized standards for safety reporting Though long considered a top priority by regulators, the process to standardize and harmonize the submission of individual case safety reports (ICSRs) has been slow. Periodic Benefit-Risk Evaluation Reports ).
FACT: A recent study conducted by the Grocery Manufacturer’s Association states that OVER 80% of packaged foods eaten in the U.S. A better definition of a G.M.O. Did you know that DANGEROUS ingredients can be found in a majority of the food items you purchase from a grocery store or any of your favorite restaurants?
The History of the ICH Q5A(R1) Guideline The International Council on Harmonization (ICH) Q5A(R1) Guideline, Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin , was first adopted by regulators in 1999. This bulk product contains both the final product (e.g., For the U.S.,
Consequently, the 1989 asbestos regulation only bans new uses of asbestos in products that would be initiated “for the first time” after 1989 and flooring felt, rollboard, and corrugated, commercial, or specialty paper. However, the majority of this ban was overturned by Corrosion Proof Fittings v. EPA, 947 F.2d 2d 1201 (5th Cir.
Guardant Vice President VICTORIA RAYMOND responded that the study was intended to demonstrate CRC sensitivity and specificity in the average risk screening population – as expected in an FDA package – and “was designed in that way, and so repeat testing was not part of the study design.” Adherence and access were a key point for the panel.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.












































Let's personalize your content