This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
However, as we note in that post, the design, timing of initiation, and timely conduct of confirmatory trials are also important considerations in FDAs determination of whether accelerated approval is appropriate. This blog post focuses on interpreting these new authorities with respect to timely conduct of confirmatory trials.
Food and Drug Administration (FDA) plays a pivotal role in fostering the development of treatments for rare diseases through its Orphan Products Grants Program. Each year, FDA selects a limited number of clinical trials to fund to help sponsors pursue development of medical products for rare diseases and advance their field.
Regulatory Guidance for Oligonucleotide Bioanalysis in DrugDevelopment pmjackson Wed, 02/19/2025 - 21:30 The unique physicochemical properties of oligonucleotides require the use of specialized bioanalytical approaches, with key considerations including selectivity and specificity, sensitivity, stability, and matrix effects.
Patients are the backbone of clinical trials, playing an essential role in the drugdevelopment process. However, patients also play a vital role in engaging directly with the FDA. It also means sponsors are less likely to waste time and resources developing programs that don’t work for patients.”
Real-world evidence (RWE) is changing clinical drugdevelopment, bridging the gap between controlled clinical trial environments and the complexities of everyday patient experiences.
Valentine — Incorporating patient and caregiver experiences into every phase of drugdevelopment has become increasingly prioritized during both development and review ( see, e.g. , previous coverage here ). At least some (if not all) of the panelists will have had experience participating in gene therapy trials.
Drugdevelopment is challenging, including not only the complexities of biomolecular drug mechanisms but also the convolutions of regulatory pathways and commercialization strategies. This includes funding large-scale clinical trials and establishing robust manufacturing/distribution networks.
Importantly, the Hub is intended to establish a new model within FDA, which leverages cross-Agency expertise in providing guidance and conducting reviews for products for rare disease populations. By Sarah Wicks & James E. Valentine & Frank J. those reviewed by the CDER Division of Rare Diseases and Medical Genetics).
Rigorous procedures to ensure that drugs are effective and safe. Regulatory bodies such as the FDA oversee clinical trials to ensure that studies’ design, conduction, analysis, and reporting are per established guidelines and laws.
Food and Drug Administration (FDA) for approval , much work remains for drugdevelopers aiming to advance treatments for pediatric populations. Reflecting Patient Diversity in NASH Trials Data from NASH clinical trials within adult populations may support clinical research into treatment for pediatric populations.
Clissold — The trio of CDER, CBER, and CDRH released a new draft guidance titled “ Use of Data Monitoring Committees in Clinical Trials ” that revises the 2006 guidance “Establishment and Operation of Clinical Trial Data Monitoring Committees” and, when final, will replace the 2006 guidance.
Snow — On September 18, 2023, FDA published an updated, final iteration of guidance for immediate implementation entitled, “ Considerations for the Conduct of Clinical Trials of Medical Products During Major Disruptions Due to Disasters and Public Health Emergencies.” By James E. Valentine & Charles D.
1] [2] It was developed by Vertex Pharmaceuticals , [5] and was approved for medical use in the United States in January 2025. [2] 2] [6] Suzetrigine is the first medication to be approved by the US Food and Drug Administration (FDA) in this new class of pain management medicines. [2] Suzetrigine is taken by mouth. [1]
The CCALC is a grassroots organization that was founded by several pharmaceutical industry members seeking clarity around the conduct of abuse and dependence potential assessments for novel drugs in development. This was in 2006, at a time when the FDA guidances on these topics had not yet been published.
On November 27,2024, FDA released a draft guidance intended to inform industry and the review staff in the Center for Drug Evaluation and Research (CDER) on how CDER views positive findings in the in vitro bacterial reverse mutation (Ames) test of a drug (active ingredient) or its metabolites and to provide recommendations on follow-up in vitro and (..)
The convergence of real-world data (RWD), technology and artificial intelligence (AI) is playing a vital role in accelerating drugdevelopment. In a recent panel discussion at DIA Global , our experts explored how these elements are reshaping clinical research and drug discovery.
Authors: Matt Cooper, PhD, Executive Director, Therapeutic Strategy Lead, Oncology; Megan Morrison, Vice President, Asia Pacific Strategy Lead Adaptive trial designs have become essential in oncology, offering a flexible and efficient approach for conducting clinical trials.
2 However, when dosed at the MTD, ADCs display improved efficacy over small molecules in oncology trials. 3D rendering of Antibody Drug Conjugate Molecules. However, a more detailed look into the clinical data suggests that there is room for further dose optimisation, and this is exemplified by the small molecule drug Sotorasib.
A new drug has entered the arsenal against Duchenne muscular dystrophy (DMD), a genetic disease that affects boys and is challenging to treat. FDA classifies it as a “nonsteroidal treatment” – not a gene therapy, but it affects gene expression. In 2023, two gene-based treatments became available.
Cato — On May 2nd, FDA released a new draft guidance with recommendations for decentralized clinical trials (DCTs) for drugs, biologics, and devices. In a DCT, trial-related activities may occur in trial participants’ homes, at local health care providers’ offices, or in local clinical laboratories. By McKenzie E.
Diversity Action Plans (DAPs) aim to improve data that the FDA receives by increasing the clinical enrollment of historically underrepresented participants. The recent draft by the FDA introduces additional guidelines for Diversity Action Plans aimed at improving diversity in clinical research.
Valentine — On November 22, 2022, FDA approved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
First, the beginning of the FDA’s START program, with the goal of accelerating the development of novel drugs and biological products for rare diseases. Selected sponsors will be able to obtain frequent advice and enhanced communication from FDA staff to address program-specific development issues within rare disease.
Sponsors using master protocols to test new drugs and therapies for treating or preventing COVID-19 should base their analyses on comparisons between control arm participants who were concurrently randomized, the FDA advised in a final guidance released yesterday. Read the guidance here: www.fdanews.com/05-17-21-COVID-19.pdf.
The year 2022 reflected a transformative path for the drugdevelopment industry. It is without a doubt that 2022 predicted change and opportunity in biopharma and biotech clinical trials in 2023. The US Food and Drug Administration (FDA) approved around 26 novel drugs in 2022.
Speakers discussed investigator, regulatory (FDA), industry, and patient perspectives during the special symposium “Challenging the Status Quo of Early Phase Clinical Trial Design: Project Optimus.” To address this challenge, the FDA Oncology Center of Excellence initiated Project Optimus. The key paradigm shift includes:
proudly announces the return of attorney Sarah Wicks to its drugdevelopment and compliance group. Sarah brings a wealth of experience and a proven track record of advising innovative drug and biologics companies through the intricate landscape of product development and commercialization.
As an undergraduate biology student, I spent some time in a TB lab working on antibiotic resistance — a growing concern for drugdevelopers. A 1994 review of 14 prospective trials and 12 case-control studies revealed that the BCG vaccine reduced the risk of TB by 50 percent. Subscribe to Asimov Press.
In today’s data-driven world, AI has become valuable and indispensable, enabling organizations to extract valuable insights from vast amounts of data, make informed decisions and drive innovation across different sectors — including drugdevelopment.
This has opened new opportunities in pharmaceutical drugdevelopment, such as the ability to evaluate large complex databases and to integrate information in useful ways. One exciting application of these technologies is the use of in silico trials in the development of novel therapies for rare diseases.
Question & Answer with Sander Vinks, PhD, PharmD, FCP, Regarding MIDD and M&S Unlocking the Power of Model-Informed DrugDevelopment (MIDD) and Modeling & Simulation (M&S) In the ever-evolving landscape of drugdevelopment, efficiency, precision, and regulatory success are paramount.
Rare diseases, therefore, present compelling opportunities for DrugDevelopment. Regulatory agencies such as the FDA have recognized these challenges and have created programs to support this research through regulatory and financial incentives. This data will enable you to move faster and do more with DrugBank.
The Animal Rule , a regulation set by the US Food and Drug Administration (FDA), applies to the development and testing of drugs and biological products intended to reduce or prevent serious or life-threatening conditions caused by exposure to lethal or permanently disabling toxic agents such as chemical, biological, radiological, or nuclear substances, (..)
Still, more than 90 percent of drug candidates fail in clinical trials, with even more that never make it to the clinical stage. Many drugs fail because they simply aren’t safe. Seal began this work after wondering if more toxicology insights could be gleaned from a drug candidate’s chemical structure. Seal S, et al.
14, 2021 /PRNewswire/ — MindMed (NEO: MMED), (OTCQB: MMEDF), (DE: MMQ), a leading psychedelic medicine biotech company today announced the addition of Robert Barrow , an accomplished pharmaceutical executive, as Chief Development Officer. We are excited to attract such top tier talent from the psychedelic drugdevelopment community.
Recently, we have seen increasing numbers of complex FIH studies in CNS-active drugs to gather comprehensive data with a wider range of doses as early as possible. jpg Tags Clinical Trials Weight 1
Five Promising Treatment Areas in Early-Phase DrugDevelopment in 2024 aasimakopoulos Wed, 04/17/2024 - 15:52 Early-phase drugdevelopment is an ever-changing landscape, as emerging science leads to new promising areas of research for the treatment of human health issues.
This process can be daunting, but understanding how to manage feedback effectively is crucial for developing and ultimately gaining approval for new therapies, especially in oncology clinical trials. An illustrative example of harmonization between agencies exists via the European Medicines Agency (EMA) and U.S.
Food and Drug Administration (FDA) recently issued a draft guidance on the development of psychedelic drugs. There has been a growing interest in using psychedelics for the treatment of medical conditions in recent years, and this guidance by the FDA is timely to support researchers investigating this use.
As we approach the end of 2023, it’s time to reflect on the past 12 months and how advances in drugdevelopment shaped the pharma and biotech industries. These shifts are a prelude to further change and progress in the clinical trial landscape in 2024. Five Predictions for the DrugDevelopment Industry in 2024 1.
Hosted by American Conference Institute, the FDA Boot Camp returns for its 40th iteration with the continued intent of providing an essential working knowledge of core FDA concepts, and real-world examples that will help you to excel in your everyday practices. The essentials of the approval process for drugs and biologics.
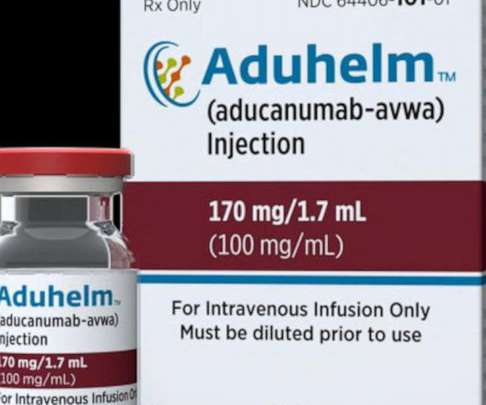
Food and Drug Administration (FDA) has approved an updated label for ADUHELM (aducanumab-avwa) injection 100 mg/mL solution. The update includes an addition to the Indications and Usage section of the label (Section 1) to emphasize the disease stages studied in the clinical trials, as seen below ( italics to note updated language).
Food and Drug Administration (FDA) announced its acceptance of the Biologics License Application (BLA) for exa-cel. In recognition of the unmet need and medical urgency for innovative therapies in the sickle cell space, the FDA granted exa-cel Priority Review, with a formal decision expected by December 8, 2023.
Sasinowski — On December 12, 2023, FDA announced the creation of a new advisory committee specifically for treatments for genetic metabolic diseases, the Genetic Metabolic Diseases Advisory Committee, or “GeMDAC.” Drugdevelopment for these conditions has unique and complex challenges, therefore few treatments are available to patients.”
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.












































Let's personalize your content