This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Results from the study that led to the FDAapproval appeared in The Lancet Neurology in April 2024 with commentary. Duvyzat reigns in HDAC activity, slowing the cascade of events leading to muscle deterioration. Adding and removing acetyls is an epigenetic change, because it leaves untouched the underlying DNA sequence.
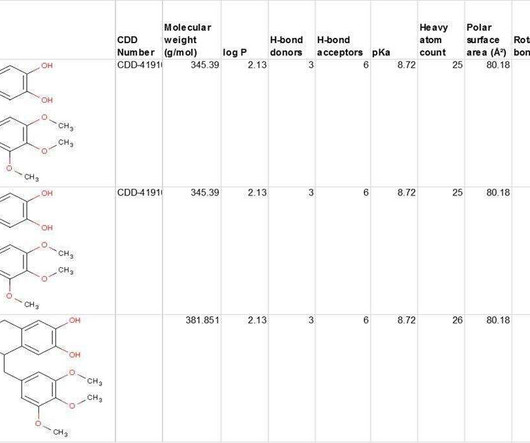
CDD Vault provides conventional SAR tables of course, but it also gives you access to data from multiple public sources for comparison with hundreds of published sources, including popular MLSMR, GlaxoSmithKline TCAMs, and FDA-Approved Re-purposed Drugs data sets.
FDAApproves Danyelza (naxitamab-gqgk) for the Treatment of Neuroblastoma. It’s very exciting to see this treatment go from being an experimental therapy used at my daughter’s bedside to now being FDAapproved. Related Articles: Danyelza (naxitamab-gqgk) FDAApproval History. NEW YORK, Nov. Source link.
FDAApproves Veklury (remdesivir) for the Treatment of COVID-19. Food and Drug Administration (FDA) has approved the antiviral drug Veklury (remdesivir) for the treatment of patients with COVID-19 requiring hospitalization. The speed and rigor with which Veklury has been developed and approved in the U.S.
Secondary VAS and pharmacokinetic (PK) endpoints and adverse events were assessed. Drug Liking and all other VAS outcomes were greatest for nabilone 3mg and 6mg, which is a currently FDA-approved medication. Three doses of lenabasum (20, 60, and 120mg) were compared to placebo, and nabilone (3 and 6mg).
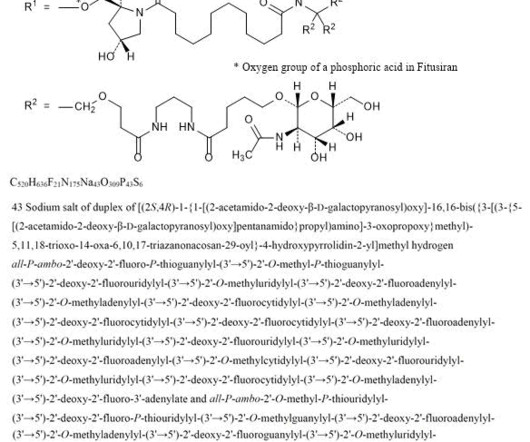
2] Fitusiran was approved for medical use in the United States in March 2025. [2] 1] [2] Adverse effects The US Food and Drug Administration prescription label for fitusiran contains a boxed warning for thrombotic events (blood clotting) and gallbladder disease (with some recipients requiring gallbladder removal). [2] 26 March 2025.
FDAApproves Klisyri (tirbanibulin) for the Treatment of Actinic Keratosis on the Face or Scalp. Food and Drug Administration (FDA) has approved Klisyri (tirbanibulin) for the topical treatment of actinic keratosis (AK) on the face or scalp. The FDAapproval of Klisyri is a significant milestone for Athenex.
Novartis today announced the US Food and Drug Administration (FDA) has granted accelerated approval for Kymriah ® (tisagenlecleucel) for the treatment of adult patients with relapsed or refractory (r/r) follicular lymphoma (FL) after two or more lines of systemic therapy.
FDAApproves Imcivree (setmelanotide) for Chronic Weight Management in Patients with Obesity Due to POMC, PCSK1 or LEPR Deficiency. With this approval, Imcivree becomes the first-ever FDAapproved therapy for these rare genetic diseases of obesity. Related Articles: Imcivree (setmelanotide) FDAApproval History.
FDAapproves Pfizer’s LITFULO™ (ritlecitinib) for adults and adolescents with severe alopecia areata Pfizer Inc. Food and Drug Administration (FDA) has approved LITFULO™ (ritlecitinib), a once-daily oral treatment, for individuals 12 years of age and older with severe alopecia areata. with placebo.
Valentine — On November 22, 2022, FDAapproved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
1 High levels of LDL-C starting at birth accelerate the event of atherosclerotic disorder , resulting in an overall increased risk of cardiovascular events, including attack and other vascular conditions, at an earlier age.2 HeFH is an inherited, genetic condition with a prevalence of 1 in 250 people worldwide.1
Forward-looking statements in this press release include, without limitation, the ability of the Company and WPD to initiate clinical trials during the first quarter of 2021, and whether the planned clinical trial will provide data to the FDA to allow an expedited pathway for development.
.
.
However, Myovant’s drug dropped the percentage of major cardiovascular events to 2.9% The secondary endpoint for increasing lifespan for patients with metastatic prostate cancer came in at 74% still alive after treatment with Orgovyx compared to 75% of those on Lupron. . compared to AbbVie’s at 6.2%.
These statements involve risks and uncertainties that could cause actual results to differ materially from those reflected in such statements, including without limitation uncertainty of success in the development and commercialization of ADUHELM; risks relating to the launch of ADUHELM, including preparedness of healthcare providers to treat patients, (..)
There were no treatment-related adverse events leading to withdrawal. “The priority review and subsequent approval of Evrysdi for babies under two months of age speaks to the urgent ongoing need for additional treatment options for babies with SMA,” said Levi Garraway, M.D.,
FDA EMERGENCY USE AUTHORIZATION PRESCRIBING INFORMATION: Do not administer Pfizer-BioNTech COVID-19 Vaccine to individuals with known history of a severe allergic reaction (e.g., Vaccination providers must report Adverse Events in accordance with the Fact Sheet to VAERS at [link] by calling 1-800-822-7967.
In order to listen to the audio webcast, interested parties can register and access the live webcast under “News/Events” through the “Investors” section of the Aurinia corporate website at www.auriniapharma.com. The Company is currently seeking FDAapproval of voclosporin for the potential treatment of LN. ABOUT AURINIA.
Teva and MedinCell Announce FDAApproval of UZEDY™ (risperidone) Extended-Release Injectable Suspension, a Long-Acting Subcutaneous Atypical Antipsychotic Injection, for the Treatment of Schizophrenia in Adults Teva Pharmaceuticals, a U.S. The primary endpoint was the frequency of all adverse events, including serious adverse events.
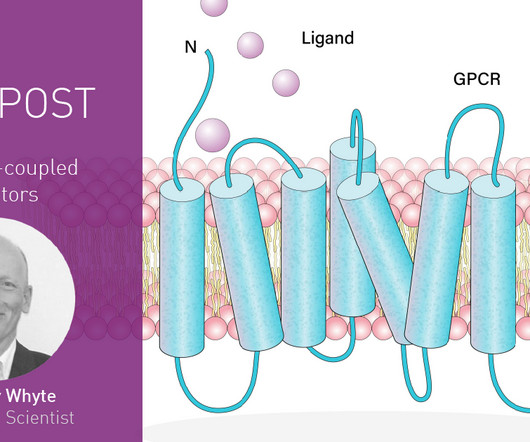
And if you need to take an FDA-approved drug, there’s around a one in three chance that it’s a drug that targets a GPCR. But what are the best ways to study this diverse group of molecules and the cascade of signaling events that ensue in the cell?
” In this head-to-head Phase IV study, patients in the Aimovig arm demonstrated a significantly lower discontinuation rate due to adverse events versus patients in the topiramate arm (10.6% Additional study treatment-related adverse events reported by ?2% versus 38.9%). .” versus 38.9%). versus 31.2%).
EST.
In order to listen to the audio webcast, interested parties can register and access the live webcast under “News/Events” through the “Investors” section of the Aurinia corporate website at www.auriniapharma.com. The Company is currently seeking FDAapproval of voclosporin for the potential treatment of lupus nephritis (LN).
HPM is ably represented by seasoned drug product development and authorization attorney—and former FDA counsel— Deborah Livornese. Deb assists pharmaceutical drug companies of all sizes on regulatory requirements and strategies related to obtaining FDAapproval and other paths to market, as well as on post-marketing regulatory requirements.
Food and Drug Administration (FDA) blessing of VUITY ™ (pilocarpine HCl ophthalmic result)1.25 VUITY is the first and only FDA-approved eye drop to treat this common and progressive eye condition that affects 128 million Americans, nearly half of theU.S. Allergan, an AbbVie ( company, moment blazoned theU.S. adult population.
DEXTENZA is FDAapproved for the treatment of ocular inflammation and pain following ophthalmic surgery. The most common non-ocular adverse event was headache (1%). Ocular Therapeutix’s first commercial drug product, DEXTENZA, is FDA-approved for the treatment of ocular inflammation and pain following ophthalmic surgery.
Food and Drug Administration (FDA) approved Actemra ® /RoActemra ® (tocilizumab) subcutaneous injection for slowing the rate of decline in pulmonary function in adult patients with systemic sclerosis-associated interstitial lung disease (SSc-ILD), a debilitating condition with limited treatment options. 1-3 SSc affects about 2.5
of new cancer drugs tested in Phase I were likely to receive Food and Drug Administration (FDA) approval. Assessing Endpoints A final hurdle faced in oncology trials is trial endpoints, or the event(s) or outcome(s) being measured to determine whether the study intervention is safe and effective.
The treatment had been granted breakthrough therapy designation, but FDAapproval would ultimately rest on the sponsor’s ability to demonstrate clinically meaningful improvement. Key Takeaway RWD and RWE play an important role in rare diseases, where capturing relevant data to support drug development is challenging.
Amphastar’s newly approved synthetic peptide product was determined by the FDA to be bioequivalent and therapeutically equivalent to Eli Lilly’s Glucagon Emergency Kit for Low Blood Sugar, which has a recombinant DNA (rDNA)-origin. Noted products are trademarks or registered trademarks of their respective owners.
. § 156, a patent may be extended only once (even if it would be eligible for extension on more than one occasion because it applies to several FDA-approved products), and only one patent may be extended for each regulatory review period. Another case of same-day (and same-time) FDAapprovals! To that end, 35 U.S.C. § 156(c)(4)
Overall, the number of treatment-emergent adverse events reported was similar between the ponesimod and teriflunomide treated groups, and the majority were mild/moderate and did not warrant treatment discontinuation. Adverse events should be reported. vs. 9.4%), nasopharyngitis (19.3% vs. 16.8%), headache (11.5% vs. 10.4%).
BYOOVIZ™ is the first FDAapproved ophthalmology biosimilar BYOOVIZ, priced 40% lower than LUCENTIS®, provides an equally effective and more affordable treatment option to patients suffering from retinal disorders BYOOVIZ will be commercially available through major distributors across the U.S. on July 1, 2022. Biogen Inc.
The transaction has been approved by a U.S. Virtual Event. The Swiss drugmaker is an outspoken advocate for digital transformation within the pharma space, though it recently ended a partnership with Pear Therapeutics on FDA-approved reSET and reSET-O products, both software-based therapeutics for substance use disorder.
Lenacapavir was generally well-tolerated, with no serious adverse events related to study drug and no study drug discontinuations through the 14-day period, including no discontinuations due to adverse events. The most common adverse events observed were injection site reactions.
Food and Drug Administration (FDA)-approved therapies for treating PAH were primarily vasodilators, designed to overcome the imbalance between vasoactive and vasodilator mediators and to restore endothelial cell function. Historically, the available drugs and U.S.
The primary endpoint of this trial is to evaluate patients for complete histological clearance of the tumor cells within the treated BCC lesion with secondary endpoints, evaluating subjects for investigational product treatment related adverse events, as well as serious adverse events, and cutaneous skin reactions.
Forward-looking statements in this press release include, without limitation, the ability of Moleculin to receive the benefits of the Orphan Drug designation, some of which require FDAapproval of Annamycin for the Orphan Drug indication, and the ability of Annamycin to demonstrate safety and efficacy in patients.
.
The majority of treatment-related adverse events (AEs) were Grade 1-2. RYBREVANT TM (amivantamab-vmjw) received accelerated approval by the U.S. All events were Grade 1-2. 1 Disease response was evaluated using overall response rate (ORR), per Response Evaluation Criteria in Solid Tumors Version 1.1* (RECIST v1.1)
SQI intends to submit RALI-dx for EUA to FDA in late Q4 2020. The RALI-dx COVID-19 Severity Triage Test is expected to be used primarily in hospital emergency departments upon FDAapproval.
SQI intends to submit RALI- fast for EUA to FDA in late Q1 2021.
.
Source link.
Although we are disappointed by the delay in timing to bring omidubicel to patients after a potential FDAapproval, we are encouraged by the FDA’s reaction to our Phase 3 data as the pivotal trial of omidubicel achieved pre-specified primary and secondary endpoints. chief executive officer of Gamida Cell.
There are currently no FDA-approved anticoagulation therapies for pediatric patients with congenital heart disease who have undergone the Fontan procedure. Xarelto is approved in more than 130 countries, although the approved labelling, including the number of indications may differ from country to country.
In the initial 275 patients, rates of adverse events (AEs) were similar among groups. Authorized Emergency Use Casirivimab and imdevimab injection is an investigational combination therapy and has been authorized by FDA for the emergency use described above. Casirivimab and imdevimab injection is not FDAapproved for any use.
FDA-approved oral prescription medicine, 120 mg or 160 mg dependent on weight (<50 kg or ?50 i Continued approval may be contingent upon verification and description of clinical benefit in confirmatory trials. For all FDA-approved indications for ALIMTA, please see full Prescribing Information.
Data from the event-driven trial could support global authorization and approval, including in the U.S. Depending on the overall COVID-19 attack rate, interim data in the UK trial, which is also event-driven, are expected as soon as early first quarter 2021. NVX-CoV2373 (SARS-CoV-2 vaccine) FDAApproval History.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.










































Let's personalize your content