This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
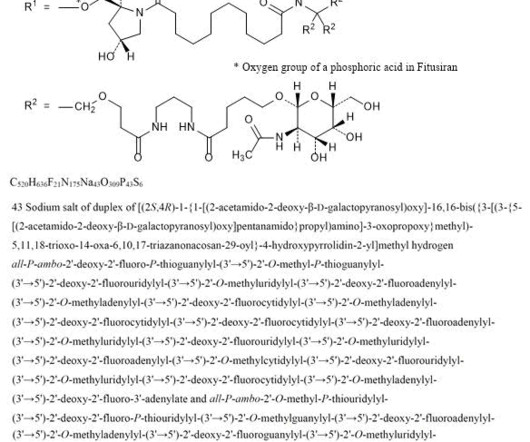
2] Fitusiran was approved for medical use in the United States in March 2025. [2] 1] [2] Adverse effects The US Food and Drug Administration prescription label for fitusiran contains a boxed warning for thrombotic events (blood clotting) and gallbladder disease (with some recipients requiring gallbladder removal). [2] 26 March 2025.
The Pfizer-BioNTech COVID-19 Vaccine has not been approved or licensed by the U.S. Food and Drug Administration (FDA), but has been authorized for emergency use by FDA under an Emergency Use Authorization (EUA) to prevent Coronavirus Disease 2019 (COVID-19) for use in individuals 16 years of age and older.
FDAApproves Danyelza (naxitamab-gqgk) for the Treatment of Neuroblastoma. It’s very exciting to see this treatment go from being an experimental therapy used at my daughter’s bedside to now being FDAapproved. Related Articles: Danyelza (naxitamab-gqgk) FDAApproval History. NEW YORK, Nov. Source link.
Valentine — On November 22, 2022, FDAapproved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
During the meeting, the FDA provided encouraging feedback regarding the Phase 3 study of omidubicel pertaining to the pre-specified primary and secondary endpoints. The FDA also recommended that Gamida Cell generate additional manufacturing-related data prior to requesting a pre-Biologics License Application (BLA) meeting.
CNS holds a worldwide exclusive license to the Berubicin chemical compound and has acquired all data and know-how from Reata Pharmaceuticals, Inc. These statements relate to future events, future expectations, plans and prospects. Any forward-looking statements contained in this press release speak only as of its date.
1, 2020 /PRNewswire/ — Sosei Group Corporation (“the Company”) (TSE: 4565) announces it has entered into a global collaboration and license agreement with Biohaven Pharmaceutical Holding Company Ltd. (“Biohaven”, NYSE: BHVN). .
TOKYO and CAMBRIDGE, England , Dec. Biohaven Forward-looking statements.
As a reminder, in Title I of the 2013 Drug Quality and Security Act (DQSA) (the Compounding Quality Act), Congress created the “outsourcing facility” FDA registration category, and set forth statutory parameters for their operation in new section 503B of the FDCA. See 21 U.S.C. 353b(a). Section II at 2. Draft Guidance III.B.2(e)
The application will be reviewed by the EMA’s Committee for Medicinal Products for Human Use (CHMP) under the centralized licensing procedure for all 27 Member States of the European Union, as well as Norway, Iceland and Liechtenstein. The most common adverse events observed were injection site reactions.
Brilacidin for UP/UPS was licensed to Alfasigma S.p.A. A Phase 2b trial of Brilacidin showed a single intravenous dose of the drug delivered comparable outcomes to a seven-day dosing regimen of the FDA-approved blockbuster daptomycin in treating Acute Bacterial Skin and Skin Structure Infection.
Source link.
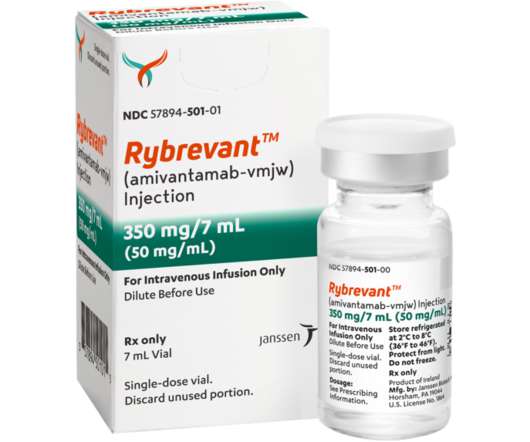
The majority of treatment-related adverse events (AEs) were Grade 1-2. RYBREVANT TM (amivantamab-vmjw) received accelerated approval by the U.S. entered into a license and collaboration agreement with Yuhan Corporation for the development of lazertinib. All events were Grade 1-2. as the primary endpoint.
Scott and colleagues focused on six genes that encode potential drug targets licensed or in development by GlaxoSmithKline for the treatment of obesity or diabetes. That’s critical considering that just 1 in 10 drug candidates entering human clinical trials successfully goes on to receive FDAapproval [5]. Novo Nordisk.
2 This is of particular importance to PNH, where one of the disease hallmarks is the mutation of the PIGA gene. [1] Research In a clinical study with twelve participants, iptacopan as a single drug led to the normalization of hemolytic markers in most patients, and no serious adverse events occurred during the 12-week study. [5]
Food and Drug Administration (FDA) has accepted for review the supplemental Biologics License Application (sBLA) for Dupixent ® (dupilumab) as an add-on treatment for children aged 6 to 11 years with uncontrolled moderate-to-severe asthma. Detailed results from this Phase 3 trial will be published later this year.
Currently, Amazon only offers special license for point of care and professional use medical products, which would preclude the individual use GenViro kit from being available on Amazon, even after FDAapproval is secured. Two of the company’s. CONTACT INFORMATION:
Decision Diagnostics Corp.
based contract manufacturing business, Benuvia Manufacturing, which has significant chemistry and formulation capabilities, including manufacturing our FDA-approved cannabinoid drug, SYNDROS ® ,” said Todd C. We look forward to supporting Radius through our U.S. Davis, executive chairman of Benuvia. Disease Highlights.
In the initial 275 patients, rates of adverse events (AEs) were similar among groups. Authorized Emergency Use Casirivimab and imdevimab injection is an investigational combination therapy and has been authorized by FDA for the emergency use described above. Casirivimab and imdevimab injection is not FDAapproved for any use.
Pfizer and BioNTech expect to file a Biologics License Application for possible full regulatory approval in 2021.
Food and Drug Administration (FDA) has authorized the emergency use of the mRNA vaccine, BNT162b2, against COVID-19 in individuals 16 years of age or older.
Under the terms of the settlement agreement, the litigation between the parties in the United States District Court for the District of New Jersey will be ended, and Lupin will have a license to sell its generic product beginning April 2033, or earlier under certain circumstances.
. “We look forward to providing an important new medicine and helping patients find the relief they so desperately seek from the varied and debilitating symptoms of this disease, contingent upon FDAapproval.” With these data, Lilly plans to submit a Biologics License Application (BLA) to the U.S. Almirall S.A.’s
–( BUSINESS WIRE )– Bristol Myers Squibb (NYSE: BMY) today announced that the Biologics License Application (BLA) for lisocabtagene maraleucel (liso-cel) for the treatment of adults with relapsed or refractory (R/R) large B-cell lymphoma after at least two prior therapies remains under review by the U.S. 1, 2021 11:59 UTC.
. “We’re excited OLUMIANT may be a potential first-in-disease medicine approved this year for adults with severe alopecia areata.” OLUMIANT, a once-daily, oral JAK inhibitor was discovered by Incyte and licensed to Lilly. It is approved in the U.S. ” About OLUMIANT®.
Founded and led for over 30 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to eight FDA-approved treatments and numerous product candidates in development, all of which were homegrown in our laboratories.
OKYO has been developing the chemerin molecule as a promising anti-inflammatory treatment for dry-eye disease (“DED”) licensed from researchers at On Target Therapeutics LLC. When Lucentis ® (Ranibizumab) received FDAapproval in late June 2006, the new macular degeneration drug was celebrated as a major medical breakthrough.
PRX-102 was well-tolerated in the study, with all adverse events being transient in nature without sequelae. The most common moderate treatment emergent adverse events were nasopharyngitis, headache and dyspnea. Protalix has licensed to Pfizer Inc. mL/min/ 1.73m 2 in males, and 86.14 nM and 13.81
Except as required by law, Moderna disclaims any intention or responsibility for updating or revising any forward-looking statements contained in this press release in the event of new information, future developments or otherwise. MRNA-1273 (SARS-CoV-2 vaccine) FDAApproval History. Source: Moderna, Inc. . Source link.
Except as required by law, Moderna disclaims any intention or responsibility for updating or revising any forward-looking statements contained in this press release in the event of new information, future developments or otherwise. MRNA-1273 (SARS-CoV-2 vaccine) FDAApproval History. Source: Moderna, Inc. . Source link.
resolving patent litigation brought in response to Teva’s Abbreviated New Drug Application, seeking approval to market a generic version of Xtampza ER prior to the expiration of Collegium’s applicable patents. FDAapproval, and customary exceptions). Reached a settlement with Teva Pharmaceutical USA, Inc.
Except as required by law, Moderna disclaims any intention or responsibility for updating or revising any forward-looking statements contained in this press release in the event of new information, future developments or otherwise. MRNA-1273 (SARS-CoV-2 vaccine) FDAApproval History. Source: Moderna, Inc. . Source link.
The most common adverse events (AEs) were cytokine release syndrome (CRS; 39%; n=19), anemia (37%; n=18), fatigue (35%; n=17), nausea (31%; n=15), pyrexia (31%; n=15) and back pain (27%; n=13). 6 months of follow-up, 83% (10 of 12 patients) have ongoing responses for up to 13 months at the time of analysis.
Serious adverse events were numerically more frequent with placebo than REGN-COV2 treatment (0.8% Regeneron does not undertake any obligation to update (publicly or otherwise) any forward-looking statement, including without limitation any financial projection or guidance, whether as a result of new information, future events, or otherwise.
Recent Events. AbbVie announced that the European Commission (EC) has approved Rinvoq (upadacitinib, 15 mg, once daily) for the treatment of adults with active psoriatic arthritis (PsA) and active ankylosing spondylitis (AS). and by AbbVie outside of the U.S. Imbruvica is jointly developed and commercialized with Janssen Biotech, Inc.
Pfizer assumes no obligation to update forward-looking statements contained in this release as the result of new information or future events or developments. BNT162b2 (SARS-CoV-2 vaccine) FDAApproval History. The information contained in this release is as of November 20, 2020. Source: Pfizer Inc. . Posted: November 2020.
Pfizer assumes no obligation to update forward-looking statements contained in this release as the result of new information or future events or developments. BNT162b2 (SARS-CoV-2 vaccine) FDAApproval History. The information contained in this release is as of November 9, 2020. Source: Pfizer, inc. Posted: November 2020.
The recipient rat was able to perform a task, which normally took eight weeks to learn, in seconds, according to comments made by program manager, Geoff Ling, at a 2015 DARPA event. Researchers used an off-the-shelf, FDA-approved machine, developed by Blackrock Neurotech ; the algorithms were the main advancement.)
ULTOMIRIS – Atypical Hemolytic Uremic Syndrome (aHUS): In September 2020 , Japan’s MHLW approved ULTOMIRIS for adults and children with aHUS. FDAapproved the ULTOMIRIS 100 mg/mL formulation for PNH and aHUS. The 2020 GAAP and non-GAAP tax rates do not benefit from one-time events that benefited the tax rates in 2019.
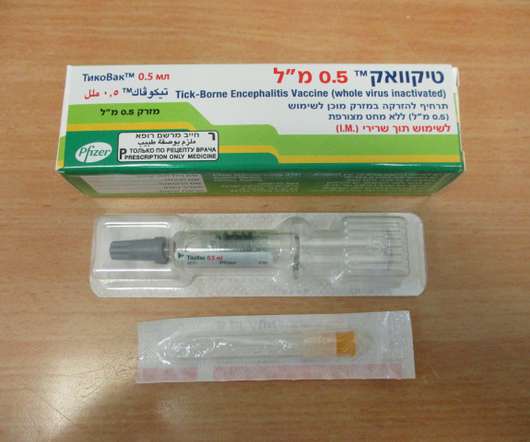
Food and Drug Administration (FDA) has approved TICOVAC (tick-borne encephalitis (TBE) vaccine) for active immunization to prevent TBE in individuals 1 year of age and older. 1 TICOVAC is the only FDA-approved vaccine to help protect U.S. Following today’s FDAapproval, the U.S. in 1-15 year olds and 98.7-100%
Trademarks are owned by or licensed to Janssen and the ViiV Healthcare group of companies. These events may have been associated with inadvertent (partial) intravenous administration and began to resolve within a few minutes after the injection.
Hepatotoxicity: Hepatic adverse events were reported. REFERENCES.
FDAApproves Zokinvy (lonafarnib) for Hutchinson-Gilford Progeria Syndrome and Processing-Deficient Progeroid Laminopathies. Eiger licensed exclusive worldwide rights to lonafarnib from Merck, known as MSD outside of the United States and Canada. Related Articles: Zokinvy (lonafarnib) FDAApproval History.
The patents cover the inhibition of soluble P-selectin that helps to prevent and/or reduce thrombotic events. Quercis’ lead drug candidate acts as an antithrombotic with significantly lower risk of adverse events than existing therapies. ZUG, Switzerland , Jan. Terms of the agreement have not been disclosed.
NASDAQ: AUPH / TSX:AUP) (“Aurinia” or the “Company”) today announced it has entered into a collaboration and license agreement with Otsuka Pharmaceutical Co., The Company is currently seeking FDAapproval of voclosporin for the potential treatment of LN. VICTORIA, British Columbia & ROCKVILLE, Md.–(
WAKIX is the first and only treatment approved by the FDA for people with excessive daytime sleepiness or cataplexy associated with narcolepsy that is not scheduled as a controlled substance by the U.S. WAKIX received FDAapproval for the treatment of excessive daytime sleepiness in adult patients with narcolepsy in August 2019.
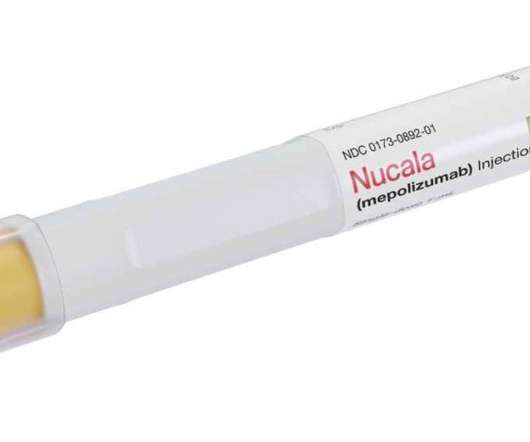
It is not currently approved for use in COPD anywhere in the world. The following information is based on the US Prescribing Information for Nucala in licensed indications only. Discontinue Nucala in the event of a hypersensitivity reaction. Important safety information. CONTRAINDICATIONS. Hypersensitivity reactions (e.g.,
Food and Drug Administration (FDA) has approved PREVNAR 20 (Pneumococcal 20-valent Conjugate Vaccine) for the prevention of invasive disease and pneumonia caused by the 20 Streptococcus pneumoniae (pneumococcus) serotypes in the vaccine in adults ages 18 years and older. Following today’s FDAapproval, the U.S.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.













































Let's personalize your content