This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Secondary VAS and pharmacokinetic (PK) endpoints and adverse events were assessed. Drug Liking and all other VAS outcomes were greatest for nabilone 3mg and 6mg, which is a currently FDA-approved medication. Three doses of lenabasum (20, 60, and 120mg) were compared to placebo, and nabilone (3 and 6mg).
of new cancer drugs tested in Phase I were likely to receive Food and Drug Administration (FDA) approval. Participants may balk at certain protocol requirements, such as extended visits for pharmacokinetic (PK) measurements. And if they do participate, they may be more likely to drop out prior to reaching a study endpoint.
The majority of treatment-related adverse events (AEs) were Grade 1-2. RYBREVANT TM (amivantamab-vmjw) received accelerated approval by the U.S. All events were Grade 1-2. 1 Disease response was evaluated using overall response rate (ORR), per Response Evaluation Criteria in Solid Tumors Version 1.1* (RECIST v1.1)
Because of its selectivity for oxycodone, the vaccine will not interfere with FDA-approved medications, including methadone, buprenorphine, naltrexone and naloxone, potentially offering a long-lasting, safe and cost-effective alternative that is complementary to standard medical intervention for opioid use disorders.
There are currently no FDA-approved anticoagulation therapies for pediatric patients with congenital heart disease who have undergone the Fontan procedure. Xarelto is approved in more than 130 countries, although the approved labelling, including the number of indications may differ from country to country.
This approach capitalizes on prior investments in R&D, mitigates risk by leveraging established safety and pharmacokinetic profiles, and accelerates the delivery of treatments to patients. The National Institutes of Health (NIH) Clinical Collection , a library of FDA-approved drugs, is widely used for HTS in drug repurposing initiatives.
In this cohort, the most common treatment-emergent adverse events of any grade (?20%) No patients in this cohort discontinued treatment due to treatment-related adverse events. ” In May 2020, Lilly’s first-in-class selective RET inhibitor Retevmo received Accelerated Approval from the U.S. Retevmo is an U.S.
PRX-102 was well-tolerated in the study, with all adverse events being transient in nature without sequelae. The most common moderate treatment emergent adverse events were nasopharyngitis, headache and dyspnea. mL/min/ 1.73m 2 in males, and 86.14 mL/min/ 1.73m 2 in females and plasma lyso-Gb 3 mean levels were 51.81 nM and 13.81
“We are pleased to continue pursuing additional neuroscience opportunities with BXCL501, targeting agitation associated with delirium, a fifth potential indication for this candidate and a condition for which there is no FDA-approved treatment,” commented Vimal Mehta, Chief Executive Officer of BTI.
For the 16-week treatment period, overall rates of adverse events (AEs) were 64% for Dupixent and 74% for placebo. Significantly improved measures of observed patient outcomes (including sleep, skin pain and health-related quality of life), as well as caregiver-reported health-related quality of life.
The most common adverse events (AEs) were cytokine release syndrome (CRS; 39%; n=19), anemia (37%; n=18), fatigue (35%; n=17), nausea (31%; n=15), pyrexia (31%; n=15) and back pain (27%; n=13). 6 months of follow-up, 83% (10 of 12 patients) have ongoing responses for up to 13 months at the time of analysis.
This includes the number of infants able to sit without support for 5 and 30 seconds, a key motor milestone not normally seen in the natural course of the disease, as well as data on event-free survival and reduced hospitalisations. SMN protein is found throughout the body and is critical for maintaining healthy motor neurons and movement.
Until recently, the FDA relied on a monograph process through which firms could bring OTC drugs to market without FDAapproval so long as it adhered to pre-set terms under the monograph. The committee also made recommendations regarding pharmacokinetic and safety assessments.
These events may have been associated with inadvertent (partial) intravenous administration and began to resolve within a few minutes after the injection.
Hepatotoxicity: Hepatic adverse events were reported. The causal relationship, mechanism, and long-term consequences of these events have not been established.
It also provided supporting pharmacokinetic data demonstrating the opioid antagonist’s safety and efficacy. . A total of 197 patients in the study arm had at least one of the composite endpoint events, compared with 312 patients in the placebo arm. New indications.
Nasdaq GILD) moment blazoned new data from an interim analysis of its ongoing, Phase2/3 single arm, open- marker study to estimate the safety, tolerability and pharmacokinetics of Veklury ® (remdesivir) in pediatric cases rehabilitated with COVID-19 with periods ranging from 28 days to lower than 18 times. Gilead Lores,Inc.
Decreases in serious adverse events, high-grade adverse events and treatment-related adverse events were observed in the second year versus the first year in both treatment arms. The most common serious adverse events were pneumonia (6.7% and 10%, respectively), nasopharyngitis (21.7% and 0%, respectively).
Read Safety and pharmacokinetics of escalating doses of neutralising monoclonal antibody CAP256V2LS administered with and without VRC07-523LS in HIV-negative women in South Africa (CAPRISA 012B): a phase 1, dose-escalation, randomised controlled trial. . Read 🏘️ Events Send me your events. Ricciardi M.J.
The FDAapproved Evrysdi in August 2020 as the first and only at home SMA treatment with proven efficacy in adults, children and infants 2 months and older. Food and Drug Administration (FDA) approved Evrysdi for the treatment of SMA in adults and children 2 months of age and older. mg/kg for Part 2.
Grade 3/4 treatment emergent adverse events (TEAEs) were reported in 63.3 Food and Drug Administration (FDA) approval for nmCRPC on February 14, 2018 and was approved for mCSPC on September 17, 2019. Ischemic cardiovascular events, including events leading to death, occurred in patients receiving ERLEADA ®.
See Warnings and Precautions in the FDA-approved full Prescribing Information for additional information on risks associated with longer-term treatment with baricitinib. It is approved in the U.S. Olumiant was recently approved in Japan for the treatment of pneumonia associated with COVID-19 in hospitalized adult patients.
In August, the FDAapproved Evrysdi for the treatment of SMA in adults and children 2 months and older. The most common adverse events (n=21) included fever (pyrexia; 71%), upper respiratory tract infection (52%), cough (33%), vomiting (33%), diarrhea (29%) and respiratory tract infection (29%).
Across 1,035 patients, there were 11 events (2.1 percent) in patients taking therapy and 36 events (7.0 Serious adverse events were reported at a similar frequency in the bamlanivimab and etesevimab together and placebo groups. volunteers to evaluate the safety, tolerability, pharmacokinetics and immunogenicity.
Metabolism of 2024 FDAapproved small molecules part 1 By Julia Shanu-Wilson Involvement of active metabolites The number of small molecules approved by the FDA in 2024 totaled 31 out of 50 NMEs with 56% (28) of these comprising small molecule therapies. Clin Pharmacokinet. What’s next? 2015 May;54(5):457-71.
months, 2,039 participants (81.8%) had any treatment-emergent adverse event (TEAE), 236 (9.5%) had a serious TEAE (SAE) and 56 (2.2%) discontinued the study due to a TEAE. Inhibition of drug transporter breast cancer resistance protein has no effect on the pharmacokinetics of major active metabolites of ozanimod. Author: Zhang.
Zidovudine showed promise against multiple HIV strains in cultured cells, and the Food and Drug Administration (FDA) approved it for human studies within five months. By 1987, the FDA licensed zidovudine after trials showed it increased survival rates.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.


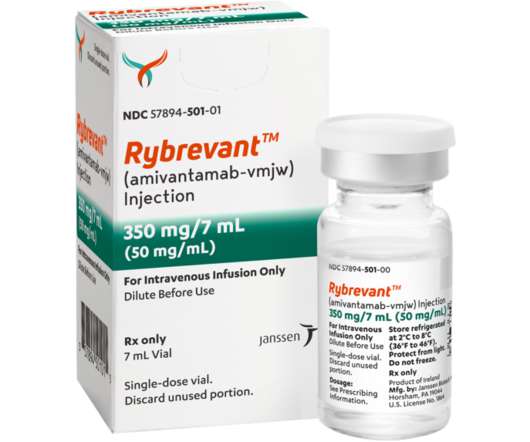
























Let's personalize your content