This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
FDA classifies it as a “nonsteroidal treatment” – not a gene therapy, but it affects gene expression. Results from the study that led to the FDAapproval appeared in The Lancet Neurology in April 2024 with commentary. Researchers have been working on developing gene therapy for DMD for decades.
Valentine — On November 22, 2022, FDAapproved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
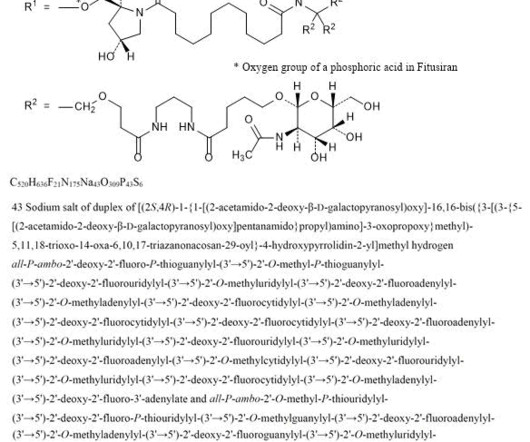
2] Fitusiran was approved for medical use in the United States in March 2025. [2] 1] [2] Adverse effects The US Food and Drug Administration prescription label for fitusiran contains a boxed warning for thrombotic events (blood clotting) and gallbladder disease (with some recipients requiring gallbladder removal). [2] 28 March 2025.
Novartis today announced the US Food and Drug Administration (FDA) has granted accelerated approval for Kymriah ® (tisagenlecleucel) for the treatment of adult patients with relapsed or refractory (r/r) follicular lymphoma (FL) after two or more lines of systemic therapy.
FDAApproves Danyelza (naxitamab-gqgk) for the Treatment of Neuroblastoma. This indication is approved under accelerated approval regulation based on overall response rate and duration of response. This indication is approved under accelerated approval based on overall response rate and duration of response.
FDAApproves Imcivree (setmelanotide) for Chronic Weight Management in Patients with Obesity Due to POMC, PCSK1 or LEPR Deficiency. Nasdaq:RYTM), a biopharmaceutical company aimed at developing and commercializing therapies for the treatment of rare genetic diseases of obesity, announced today that the U.S. BOSTON, Nov.
FDAApproves Klisyri (tirbanibulin) for the Treatment of Actinic Keratosis on the Face or Scalp. NASDAQ: ATNX), a global biopharmaceutical company dedicated to the discovery, development, and commercialization of novel therapies for the treatment of cancer and related conditions, today announced that the U.S. BUFFALO, N.Y.,
FDAApproves Veklury (remdesivir) for the Treatment of COVID-19. Food and Drug Administration (FDA) has approved the antiviral drug Veklury (remdesivir) for the treatment of patients with COVID-19 requiring hospitalization. The speed and rigor with which Veklury has been developed and approved in the U.S.
However, Myovant’s drug dropped the percentage of major cardiovascular events to 2.9% The secondary endpoint for increasing lifespan for patients with metastatic prostate cancer came in at 74% still alive after treatment with Orgovyx compared to 75% of those on Lupron. . compared to AbbVie’s at 6.2%.
Viral vectors have been crucial in transforming the gene therapy landscape due to their natural ability to infect cells. 1 In 2017, the US Food and Drug Administration (FDA) approved the first AAV-based gene replacement therapy (Luxturna), for Leber congenital amaurosis type 2.
Food and Drug Administration (FDA) has approved Repatha® (evolocumab) as an adjunct to diet and other LDL cholesterol (LDL-C)-lowering therapies for the treatment of pediatric patients aged 10 years and older with heterozygous hypercholesterolemia (HeFH) to scale back LDL-C. No new safety risks were identified.5
of women who continued on relugolix combination therapy remained responders (menstrual blood loss < 80 mL) through Week 76 compared with 15.1% of women who continued relugolix combination therapy remained responders through Week 104. of women who discontinued treatment at Week 52 (p < 0.0001). weeks after discontinuation.
The Company will initiate its trial during the first quarter of 2021 to investigate the efficacy of Berubicin in adults with GBM who have failed first-line therapy. These statements relate to future events, future expectations, plans and prospects.
.
SOURCE CNS Pharmaceuticals, Inc.
” In this head-to-head Phase IV study, patients in the Aimovig arm demonstrated a significantly lower discontinuation rate due to adverse events versus patients in the topiramate arm (10.6% Additional study treatment-related adverse events reported by ?2% versus 38.9%). .” versus 38.9%). versus 31.2%).
In order to listen to the audio webcast, interested parties can register and access the live webcast under “News/Events” through the “Investors” section of the Aurinia corporate website at www.auriniapharma.com. The Company is currently seeking FDAapproval of voclosporin for the potential treatment of LN. ABOUT AURINIA.
FDA EMERGENCY USE AUTHORIZATION PRESCRIBING INFORMATION: Do not administer Pfizer-BioNTech COVID-19 Vaccine to individuals with known history of a severe allergic reaction (e.g., Immunocompromised persons, including individuals receiving immunosuppressant therapy, may have a diminished immune response to the Pfizer-BioNTech COVID-19 Vaccine.
In the midst of the global pandemic, the analysis found no antiretroviral therapy interruptions across the entirety of the ongoing clinical development programme for long-acting cabotegravir and rilpivirine. Of those participants who transitioned back to injectables, the median duration of oral therapy was 51 days. Source: GSK .
Biogen discovers, develops and delivers worldwide innovative therapies for people living with serious neurological and neurodegenerative diseases as well as related therapeutic adjacencies. Please see full Prescribing Information including Medication Guide. About Biogen. At Biogen, our mission is clear: we are pioneers in neuroscience.
Food and Drug Administration (FDA) approved Actemra ® /RoActemra ® (tocilizumab) subcutaneous injection for slowing the rate of decline in pulmonary function in adult patients with systemic sclerosis-associated interstitial lung disease (SSc-ILD), a debilitating condition with limited treatment options. 1-3 SSc affects about 2.5
Food and Drug Administration (FDA)-approvedtherapies for treating PAH were primarily vasodilators, designed to overcome the imbalance between vasoactive and vasodilator mediators and to restore endothelial cell function. Historically, the available drugs and U.S. None offers a cure for PAH.
.
In order to listen to the audio webcast, interested parties can register and access the live webcast under “News/Events” through the “Investors” section of the Aurinia corporate website at www.auriniapharma.com. The Company is currently seeking FDAapproval of voclosporin for the potential treatment of lupus nephritis (LN).
Teva and MedinCell Announce FDAApproval of UZEDY™ (risperidone) Extended-Release Injectable Suspension, a Long-Acting Subcutaneous Atypical Antipsychotic Injection, for the Treatment of Schizophrenia in Adults Teva Pharmaceuticals, a U.S. We are committed to supporting patients through innovative therapy options.
of new cancer drugs tested in Phase I were likely to receive Food and Drug Administration (FDA) approval. cell and gene therapies), with other therapeutic areas then pushing it further. Patients may be wary of joining an experimental trial and taking on those potential risks when approvedtherapies are already available.
Nasdaq: GMDA), an advanced cell therapy company committed to cures for blood cancers and serious hematologic diseases, today announced that the company conducted a Type B Meeting for omidubicel with the U.S. Food and Drug Administration (FDA) on Friday, December 11, 2020. BOSTON–( BUSINESS WIRE )– Gamida Cell Ltd.
Novartis will present 48 abstracts from its leading MS portfolio, including new data on recently FDA-approved Kesimpta ® (ofatumumab) — the first and only self-administered, targeted B-cell therapy for relapsing forms of MS (RMS )— Mayzent ® (siponimod) and Gilenya ® (fingolimod) .
BYOOVIZ™ is the first FDAapproved ophthalmology biosimilar BYOOVIZ, priced 40% lower than LUCENTIS®, provides an equally effective and more affordable treatment option to patients suffering from retinal disorders BYOOVIZ will be commercially available through major distributors across the U.S. on July 1, 2022. Biogen Inc.
DEXTENZA is FDAapproved for the treatment of ocular inflammation and pain following ophthalmic surgery. The most common non-ocular adverse event was headache (1%). Ocular Therapeutix’s first commercial drug product, DEXTENZA, is FDA-approved for the treatment of ocular inflammation and pain following ophthalmic surgery.
156, a patent may be extended only once (even if it would be eligible for extension on more than one occasion because it applies to several FDA-approved products), and only one patent may be extended for each regulatory review period. PTE applications were submitted to FDA for each of U.S. To that end, 35 U.S.C. § 156(c)(4)
Lenacapavir was generally well-tolerated, with no serious adverse events related to study drug and no study drug discontinuations through the 14-day period, including no discontinuations due to adverse events. The most common adverse events observed were injection site reactions.
Overall, the number of treatment-emergent adverse events reported was similar between the ponesimod and teriflunomide treated groups, and the majority were mild/moderate and did not warrant treatment discontinuation. Adverse events should be reported. vs. 9.4%), nasopharyngitis (19.3% vs. 16.8%), headache (11.5% vs. 10.4%).
Current guidelines are limited and recommend treating pediatric patients with or at risk for reoccurrence of blood clots with standard anticoagulation therapy which requires injections, dietary restrictions, and regular laboratory monitoring. Study The randomized, open-label phase III EINSTEIN-Jr.
Retevmo was approved under the FDA’s Accelerated Approval regulations based on the LIBRETTO-001 Phase 1/2 trial’s endpoints of overall response rate (ORR) and duration of response (DoR). Session Title: Targeted Therapy and Ovarian Cancer Trials. Abstract Number: CT011. Retevmo is an U.S. About Sintilimab.
The treatment had been granted breakthrough therapy designation, but FDAapproval would ultimately rest on the sponsor’s ability to demonstrate clinically meaningful improvement. Key Takeaway RWD and RWE play an important role in rare diseases, where capturing relevant data to support drug development is challenging.
In the initial 275 patients, rates of adverse events (AEs) were similar among groups. If the therapy proves safe and effective in clinical trials and regulatory approvals are granted, Regeneron will manufacture and distribute it in the U.S. , Casirivimab and imdevimab injection is not FDAapproved for any use.
This allows for early detection of adverse events and treatment optimization based on objective real-world evidence. This may enable the development of highly targeted therapies and individualized treatment plans, maximizing therapeutic efficacy while minimizing the risk of adverse side effects.
It is not known whether CD24 on cancer cells has a unique epitope that can be specifically targeted for cancer therapy. Combination therapy is the best approach in immunotherapy. Our extensive data showed that CD24 expressed on normal cells and cancer cells can be differentiated with our novel anti-CD24 antibody, ONC-781.
3] While MET inhibitors have recently received accelerated approval in this setting in some regions, the vast majority of patients eventually acquire resistance to these therapies, thus underscoring the need for new treatment options. [4] The majority of treatment-related adverse events (AEs) were Grade 1-2. 4] , [5] , [6].
SAMI is a renal replacement therapy (RRT) machine manufactured and commercialized by Spectral’s wholly-owned subsidiary, Dialco Medical Inc. ( “Dialco” ). The paper describes the first month of using SAMI as part of UMMC’s PIRRT (Prolonged Intermittent Renal Replacement Therapy) program throughout April 2020.
Among others, NGS has led to the identification of disease-causing variants and novel drug targets and an improved understanding of complex biological events, e.g., the heterogeneity of tumors. Drug development The application of NGS has fueled the development of targeted therapies, precision medicine , and improved diagnostic methods.
In this cohort, the most common treatment-emergent adverse events of any grade (?20%) No patients in this cohort discontinued treatment due to treatment-related adverse events. ” In May 2020, Lilly’s first-in-class selective RET inhibitor Retevmo received Accelerated Approval from the U.S. . Retevmo is an U.S.
The primary endpoint of this trial is to evaluate patients for complete histological clearance of the tumor cells within the treated BCC lesion with secondary endpoints, evaluating subjects for investigational product treatment related adverse events, as well as serious adverse events, and cutaneous skin reactions.
.
Moleculin recently announced that the FDA had allowed its request for Investigational New Drug (IND) status for Annamycin, allowing Moleculin to begin a Phase 1B /2 clinical trial in the US for patients with soft tissue sarcoma (STS) that has metastasized to the lungs after first-line therapy for their disease.
limited efficacy of experimental therapy) Change to the research resulting in increased burden or discomfort New alternative therapy availability (e.g., FDAapproval of a new drug for the condition under study) Impact of participation on alternative therapies (e.g.,
based contract manufacturing business, Benuvia Manufacturing, which has significant chemistry and formulation capabilities, including manufacturing our FDA-approved cannabinoid drug, SYNDROS ® ,” said Todd C. There are currently no approvedtherapies to treat this disorder’s hyperphagia, anxiety, or metabolic aspects.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.










































Let's personalize your content