This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
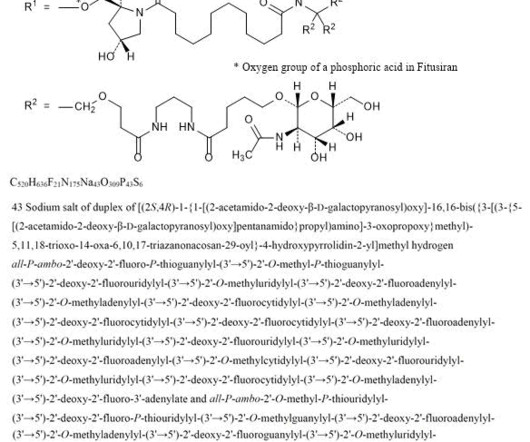
2] Fitusiran was approved for medical use in the United States in March 2025. [2] 1] [2] Adverse effects The US Food and Drug Administration prescription label for fitusiran contains a boxed warning for thrombotic events (blood clotting) and gallbladder disease (with some recipients requiring gallbladder removal). [2] 26 March 2025.
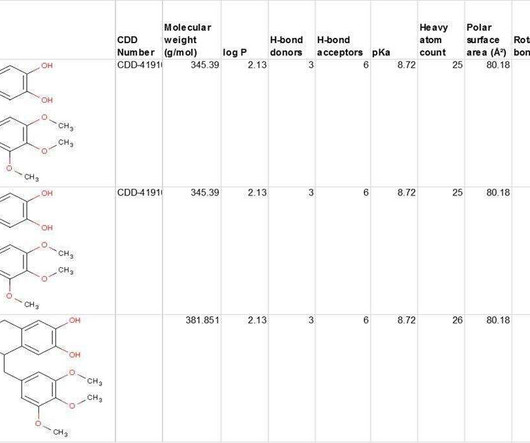
CDD Vault provides conventional SAR tables of course, but it also gives you access to data from multiple public sources for comparison with hundreds of published sources, including popular MLSMR, GlaxoSmithKline TCAMs, and FDA-Approved Re-purposed Drugs data sets. Learn more about the benefits of CDD Vault and get your free trial:
The New Drug Duvyzat (givinostat), a type of drug called an HDAC inhibitor, has been in clinical trials to treat cancers and other disorders of the blood, Crohn’s disease, and a form of juvenile arthritis. Results from the study that led to the FDAapproval appeared in The Lancet Neurology in April 2024 with commentary.
Food and Drug Administration (FDA)-approved therapies for treating PAH were primarily vasodilators, designed to overcome the imbalance between vasoactive and vasodilator mediators and to restore endothelial cell function. Their early definition — as well as plans for recording and tracking — is a major factor in a trial’s success.
FDAApproves Veklury (remdesivir) for the Treatment of COVID-19. Food and Drug Administration (FDA) has approved the antiviral drug Veklury (remdesivir) for the treatment of patients with COVID-19 requiring hospitalization. The speed and rigor with which Veklury has been developed and approved in the U.S.
FDAApproves Danyelza (naxitamab-gqgk) for the Treatment of Neuroblastoma. This indication is approved under accelerated approval regulation based on overall response rate and duration of response. The FDA granted approval under the accelerated approval regulation. NEW YORK, Nov. Contraindications.
Novartis today announced the US Food and Drug Administration (FDA) has granted accelerated approval for Kymriah ® (tisagenlecleucel) for the treatment of adult patients with relapsed or refractory (r/r) follicular lymphoma (FL) after two or more lines of systemic therapy.
Valentine — On November 22, 2022, FDAapproved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
FDAApproves Imcivree (setmelanotide) for Chronic Weight Management in Patients with Obesity Due to POMC, PCSK1 or LEPR Deficiency. With this approval, Imcivree becomes the first-ever FDAapproved therapy for these rare genetic diseases of obesity. BOSTON, Nov. in the first quarter of 2021.
The update includes an addition to the Indications and Usage section of the label (Section 1) to emphasize the disease stages studied in the clinical trials, as seen below ( italics to note updated language). Continued approval for this indication may be contingent upon verification of clinical benefit in confirmatory trial(s).
of new cancer drugs tested in Phase I were likely to receive Food and Drug Administration (FDA) approval. Innovation Organizations conducting oncology clinical trials face challenges distinct from the rest of the research community. Typical clinical development timelines for anticancer drugs average an estimated 6.7
FDAapproves Pfizer’s LITFULO™ (ritlecitinib) for adults and adolescents with severe alopecia areata Pfizer Inc. Food and Drug Administration (FDA) has approved LITFULO™ (ritlecitinib), a once-daily oral treatment, for individuals 12 years of age and older with severe alopecia areata. with placebo.
FDAApproves Klisyri (tirbanibulin) for the Treatment of Actinic Keratosis on the Face or Scalp. Food and Drug Administration (FDA) has approved Klisyri (tirbanibulin) for the topical treatment of actinic keratosis (AK) on the face or scalp. The FDAapproval of Klisyri is a significant milestone for Athenex.
The Company will initiate its trial during the first quarter of 2021 to investigate the efficacy of Berubicin in adults with GBM who have failed first-line therapy. and 2 trials planned by our sublicensee WPD in Poland. . and 2 trials planned by our sublicensee WPD in Poland.
About CNS Pharmaceuticals, Inc.
The Phase III study was conducted as an open-label trial with patients receiving either an Orgovyx pill daily or injections of Abbvie’s Lupron as an injection every three months. In the trial, Myovant’s drug outperformed Abbvie’s by suppressing testosterone to castration levels in 96.7%, compared to their competitor’s 88.8%. . “We
1 High levels of LDL-C starting at birth accelerate the event of atherosclerotic disorder , resulting in an overall increased risk of cardiovascular events, including attack and other vascular conditions, at an earlier age.2 HeFH is an inherited, genetic condition with a prevalence of 1 in 250 people worldwide.1
Among others, NGS has led to the identification of disease-causing variants and novel drug targets and an improved understanding of complex biological events, e.g., the heterogeneity of tumors. In addition, NGS facilitates transcriptomics , epigenomics, metagenomics, and other omics studies. Fountzilas et al.
The first patients have been enrolled in a phase 1 randomized placebo-controlled clinical trial to study a therapeutic vaccine for opioid use disorder developed by researchers at the University of Minnesota Medical School.
regulators to seek approval of our COVID-19 vaccine based on our pivotal Phase 3 trial and follow-up data.”. This includes the most recent analyses from the pivotal Phase 3 clinical trial, where the vaccine’s efficacy and favorable safety profile were observed up to six months after the second dose.
Results reinforce well-established safety profile of Dupixent – the first ever biologic medicine for atopic dermatitis currently approved for patients as young 6 years old. The trial demonstrated similar safety results to the known safety profile of Dupixent in atopic dermatitis. Yancopoulos, M.D., In 2016, the U.S.
There were no treatment-related adverse events leading to withdrawal. “The priority review and subsequent approval of Evrysdi for babies under two months of age speaks to the urgent ongoing need for additional treatment options for babies with SMA,” said Levi Garraway, M.D.,
Fast Track designation is well-timed, as we anticipate starting our Phase 2 clinical trial in hospitalized COVID-19 patients this month, and should help bring Brilacidin to patients faster in these dire times.”. Brilacidin, a versatile compound with broad therapeutic potential, is in a new chemical class called defensin-mimetics.
Adjuvanted S-Trimer COVID-19 vaccine candidates demonstrated favorable safety and tolerability profiles and strong neutralizing immune responses in a phase 1 trial.
Clover plans to initiate a global phase 2/3 trial in the first half of 2021 with an interim analysis for vaccine efficacy potentially in the middle of 2021.
Teva and MedinCell Announce FDAApproval of UZEDY™ (risperidone) Extended-Release Injectable Suspension, a Long-Acting Subcutaneous Atypical Antipsychotic Injection, for the Treatment of Schizophrenia in Adults Teva Pharmaceuticals, a U.S. The primary endpoint was the frequency of all adverse events, including serious adverse events.
MBA, trial investigator and managing medical director at Charité Universitätsmedizin in Berlin. ” In this head-to-head Phase IV study, patients in the Aimovig arm demonstrated a significantly lower discontinuation rate due to adverse events versus patients in the topiramate arm (10.6% versus 38.9%). . versus 38.9%). .”
Hosts a Panel Discussion on Clinical Trials with Neil Solomons, M.D., EST.
In order to listen to the audio webcast, interested parties can register and access the live webcast under “News/Events” through the “Investors” section of the Aurinia corporate website at www.auriniapharma.com. EST;
Scott Gottlieb, M.D. EST; and.
Food and Drug Administration (FDA) approved Actemra ® /RoActemra ® (tocilizumab) subcutaneous injection for slowing the rate of decline in pulmonary function in adult patients with systemic sclerosis-associated interstitial lung disease (SSc-ILD), a debilitating condition with limited treatment options. About the focuSSced Trial.
DEXTENZA is FDAapproved for the treatment of ocular inflammation and pain following ophthalmic surgery. The safety of DEXTENZA was assessed in three Phase 3 clinical trials and a Phase 2 clinical trial. The most common non-ocular adverse event was headache (1%). About DEXTENZA. About Ocular Therapeutix, Inc.
Clinical trials for ultra-rare diseases can be particularly challenging to mount due to small, geographically-dispersed patient populations. For such trials, the US Food and Drug Administration (FDA) may allow the use of credible real-world data (RWD) and real-world evidence (RWE) in lieu of data collected in a Phase 3 trial.
Phase III BRIDGE open-label, switch-over clinical trial met key objectives for safety and efficacy.
PRX-102 was well-tolerated in the study, with all adverse events being transient in nature without sequelae. The most common moderate treatment emergent adverse events were nasopharyngitis, headache and dyspnea.
The new findings from the Phase 3 clinical trials (ADvocate 1 and 2) showed eight out of ten patients who achieved clinical response (EASI-75*) with lebrikizumab monotherapy at 16 weeks maintained skin clearance at one year of treatment with the once every two weeks or four weeks regimen. Almirall S.A.’s Source link: [link].
Retevmo was approved under the FDA’s Accelerated Approval regulations based on the LIBRETTO-001 Phase 1/2 trial’s endpoints of overall response rate (ORR) and duration of response (DoR). LY3484356, an oral SERD, is currently being studied in a Phase 1/2 clinical trial. Pipeline Highlights.
The trial is comprised of three dose escalation cohorts ranging from 30 ?g g with five patients in each group, and a total of 15 patients will be enrolled in the trial. ” The Company expects to report initial clinical data from the trial in 2021. 1 and COX-2. the founder, President and CEO of Sirnaomics.
Novavax expects to begin its pivotal Phase 3 clinical trial in the United States and Mexico by the end of November. Data from the event-driven trial could support global authorization and approval, including in the U.S. NVX-CoV2373 (SARS-CoV-2 vaccine) FDAApproval History. and globally.”. About NVX-CoV2373.
The Company plans to initiate a Phase 2 trial within the next several months. “We An exploratory endpoint of this trial will be to determine the overall clinical improvement after drug administration using the Clinical Global Impression – Improvement Scale (“CGI-I”).
Following review of the application, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency based its positive opinion on data from a rolling review of trial data from the primary analysis of the Phase III programme led by the University of Oxford. AZD1222 (SARS-CoV-2 vaccine) FDAApproval History.
Following review of the application, the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency based its positive opinion on data from a rolling review of trial data from the primary analysis of the Phase III programme led by the University of Oxford. AZD1222 (SARS-CoV-2 vaccine) FDAApproval History.
Overall, the number of treatment-emergent adverse events reported was similar between the ponesimod and teriflunomide treated groups, and the majority were mild/moderate and did not warrant treatment discontinuation. Adverse events should be reported. vs. 9.4%), nasopharyngitis (19.3% vs. 16.8%), headache (11.5% vs. 10.4%).
The company believes the clinical comparability requirement will be met if the time to neutrophil engraftment in patients from the company’s ongoing expanded access program (EAP) using omidubicel produced at Gamida Cell’s planned commercial manufacturing sites is consistent with the results achieved in the Phase 3 clinical trial.
This regimen was shown in clinical trials to be safe and effective at preventing symptomatic COVID-19, with no severe cases and no hospitalisations more than 14 days after the second dose. We would like to thank our many colleagues at AstraZeneca, Oxford University, the UK government and the tens of thousands of clinical trial participants.”.
Today’s data, involving an additional 524 patients from the ongoing Phase 2/3 trial, provides definitive final virology results and meets the clinical endpoint of reducing medical visits. REGN-COV2 was well tolerated in the trial. Serious adverse events were numerically more frequent with placebo than REGN-COV2 treatment (0.8%
RAD011 is a pivotal-trial ready synthetic cannabidiol oral solution with potential utilization in multiple endocrine and metabolic orphan diseases.
Prader-Willi syndrome (“PWS”) will be the initial indication, which has been granted Orphan Drug and Fast Track Designation by the FDA.
Disease Highlights.
.
Moleculin recently announced that the FDA had allowed its request for Investigational New Drug (IND) status for Annamycin, allowing Moleculin to begin a Phase 1B /2 clinical trial in the US for patients with soft tissue sarcoma (STS) that has metastasized to the lungs after first-line therapy for their disease.
I am pleased with the clinical progress we are making with this program and we are planning to present interim data from the ongoing Phase 1/2 dose escalation trial later this year.”. About the Phase 1/2 Trial for HPN217. Dose escalation for HPN217 in the Phase 1/2 clinical trial is progressing rapidly.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.










































Let's personalize your content