This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
However, as we note in that post, the design, timing of initiation, and timely conduct of confirmatory trials are also important considerations in FDAs determination of whether accelerated approval is appropriate. This blog post focuses on interpreting these new authorities with respect to timely conduct of confirmatory trials.
A surrogate endpoint is a marker used in clinical trials as a substitute for a direct clinical outcome. For example, transcriptomic processes are showing the potential to identify and track failures in gene expression and gene regulation of amyloid and tau-related biomarkers, understood as precursors to the onset of Alzheimers disease (AD).
In an era where clinical trials are increasingly global, it’s more imperative than ever to leverage international expertise. Data and safety monitoring boards (DSMBs), also known as data monitoring committees (DMCs), play a critical role in overseeing a clinical trial’s safety and efficacy.
Tobolowsky & Véronique Li, Senior Medical Device Regulation Expert & David B. Moreover, DMCs are being used in trials of modest size and in the context of increased globalization of medical product development. In another update, the recent draft guidance added “entities reviewing safety data” and adaptation committees.
New privacy regulations seem to form every few months, especially with individual U.S. states adopting their own privacy regulations (e.g., Endpoint adjudication committees (EACs), also called clinical event committees (CECs), receive potentially identifiable research data from all over the world.
Their early definition — as well as plans for recording and tracking — is a major factor in a trial’s success. Their early definition — as well as plans for recording and tracking — is a major factor in a trial’s success.
The conference is billed as the premier event to provide folks with a roadmap to navigate the difficult terrain of FDA regulatory law. This is achieved through firsthand insights, real-world examples, and case studies from FDA-regulated products. You can access the conference brochure and sign up for the event here.
At the recent World Orphan Drug Congresses (WODC) in Europe and the United States, Worldwide Clinical Trials Derek Ansel , Vice President, Therapeutic Strategy Lead, Rare Disease, facilitated roundtable discussions to explore the operational challenges and ethical barriers surrounding genetic testing.
By harnessing the full range of innovative technologies and taking advantage of an FSP partners extensive skills and experience, sponsors are able to bring their therapies to market more quickly and within budget even in the face of complicated global regulations and widely fluctuating workloads.
For many patients, involvement in oncology clinical trials represents a last hope for an effective therapy. This is why oncology trials must be built around patient needs, and sponsors need to balance the complexity of oncology trials with a patient-centric mindset. Many of these patients’ conditions are disabling.
1 Regulators invest significant consideration balancing quality-of-life measures with overall survival when assessing novel oncology treatments. 2 However, when dosed at the MTD, ADCs display improved efficacy over small molecules in oncology trials. 3D rendering of Antibody Drug Conjugate Molecules.
Explore how emerging trends in clinical trial investigations are bringing patients closer to medicine making them more patient-centric.
The patient experience is an important consideration in trial design.
Innovative ways regulators are implementing new clinical practices to measure the patient perspective.
Assessing and reporting adverse events (AEs) in clinical trials is critical to ensuring the study is as safe as possible and the participants have the most up to date information so they can decide whether to continue their participation in the research study. regulations. Who Should Assess AEs in Clinical Trials?
There has also been an increase in government and regulatory support for CGT trials, in conjunction with an increase in investments for these products to get to market. They also need to have the medical competencies to manage adverse events often associated with these therapies.
Contract research organizations (CROs) and pharmaceutical companies can leverage these cutting-edge technologies to streamline clinical trials and introduce automation in drug discovery. As clinical trials grow in complexity, the volume of data being gathered and utilized for these studies is expanding.
But what does “working” really mean when efficacy means something subjective, like quality of life, reduced psychiatric events, or less symptoms? In most cases, the results you are really looking for and trying to prove in a trial can be complex.
Brazil’s National Agency for Sanitary Surveillance (ANVISA) has suspended Beijing, China-based Sinovac’s phase 3 trial of its COVID-19 vaccine, CoronaVac, halting the study Tuesday to evaluate a serious adverse event that occurred. Source link.
Clinical trials are needed to determine whether a medical device can produce sufficient proof of its efficacy. However, medical device sponsors and developers are limited by the complex regulatory processes and the high costs associated with the design of running clinical trials for their products.
In the absence of a clinical trial result or FDA label to point to, how does one create the case and target product profile (TPP) around a new target? If adverse events are anticipated, it is important to understand gene dosage for such an effect (e.g., One example of such a genetics exercise is represented in Fig.
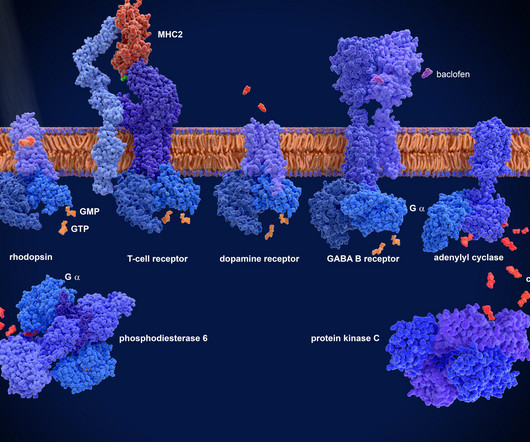
Involved in various physiological processes, such as vision, taste, smell, immune response and neurotransmission, GPCRs are activated by various molecules including hormones, neurotransmitters and environmental stimuli, which trigger a cascade of cellular events that help regulate bodily functions.
Managing clinical trial budgets efficiently is necessary for the success and sustainability of clinical research sites. Effective budget management not only ensures trials are financially viable but also maximizes return on investment (ROI). the impact and value of the data produced).
Neuropsychiatric disorders, affecting millions worldwide, disrupt the brain’s intricate processes of mood regulation, cognition and behaviour. Event-driven pharmacology Donello highlights the growing recognition of synaptic plasticity’s crucial role in the biology of depression.
Clinical trials continue to move closer to patients. Improvements to data collection technologies and processes, a robust high-speed internet infrastructure, and pressure from regulators to make research more inclusive are converging, making exclusively site-based study designs increasingly rare. Luckily, there is one solution.
What we expect European regulators to do in May 2024 In this recurring feature, AgencyIQ, through public data and previous analysis, determines what European medicine and device regulators will likely do in the month ahead, including key deadlines, meetings, events, planned regulations, comment periods, and more.
This Draft Guidance focuses on the use of patient-level data from other clinical trials or from RWD sources. For example, objective response rate is often used as a single-arm trial endpoint in oncology given the established understanding that tumor shrinkage rarely occurs without an intervention.”
Johnson & Johnson Posts Interim Results from Phase 1/2a Clinical Trial of its Janssen COVID-19 Vaccine Candidate. Johnson & Johnson Posts Interim Results from Phase 1/2a Clinical Trial of its Janssen COVID-19 Vaccine Candidate. The full set of results will be published once the complete Phase 1/2a trial data are available.
AstraZeneca’s COVID-19 vaccine candidate is facing another potential delay as the company plans to undertake a new global clinical trial to confirm the vaccine’s 90 percent efficacy based on a one-and-a-half-dose regimen. An additional trial could further delay AZ’s efforts to earn an EUA in the U.S., James Miessler. Source link.
Among others, NGS has led to the identification of disease-causing variants and novel drug targets and an improved understanding of complex biological events, e.g., the heterogeneity of tumors. It has become a fundamental tool for researchers to explore the complexities of genetic information and conduct genetic-informed drug development.
The Alliance fosters pre-competitive collaboration with members working together as equals on projects that will benefit the industry as a whole—whether it is identifying the root causes of inefficiencies, working with regulators to adopt new standards, or helping researchers implement AI effectively—and more! The
However, with this most recent approval, FDA did not leave the question of durability as something to be answered postapproval, which signals to us that this issue looms large in FDA’s preapproval regulation of gene therapies. We can see this evolution for requiring longer-term follow-up play out with this most recent approval of Hemgenix.
BRAF and downstream mitogen-activated extracellular signal-regulated kinase (MEK) inhibitors are combined to treat BRAF-mutated melanoma, for example. Inhibiting KDM5A, a regulator of cell proliferation, poses another interesting approach.
In other words, the FDA also regulates whether investigational products may be manufactured, shipped, and administered to human subjects who participate in clinical investigations. . There are three types of amendments specified in these regulations: . The sponsor of the investigation submits an IND to the FDA; . 21 CFR 312.40(a)
COVID-19 vaccine developers are faced with an ethical quandary — whether to let phase 3 trial participants become “unblinded” and to receive authorized vaccines as they become available, which could hinder the collection of meaningful trial data. Pfizer, which has received emergency approvals in the U.S.
device regulation timelines To kick off 2024, the British device regulator offered its medical device and IVD plans for this year and next, promising public action on the post-market surveillance regulation by mid-2024 and on the core regulations in late 2024 or early 2025. New roadmap sets out U.K.
September 23, 2020 – Johnson & Johnson (NYSE: JNJ) (the Company) today announced the launch of its large-scale, pivotal, multi-country Phase 3 trial (ENSEMBLE) for its COVID-19 vaccine candidate, JNJ-78436735, being developed by its Janssen Pharmaceutical Companies. NEW BRUNSWICK, N.J., In the U.S.,
A regulatory binder is essential for managing clinical trial documents, ensuring regulatory compliance, and facilitating audits. It organizes critical documents; provides easy access for trial monitors, auditors, and regulatory authorities; and serves as a reference for the research team.
LYON, France–( BUSINESS WIRE )– MaaT Pharma announced today that it treated its first patient in a Phase 1 clinical trial to evaluate the safety and tolerability of MaaT033, a capsule formulation of the company’s lead biotherapeutic, MaaT013, characterized by high microbial species diversity and richness.
The Phase 1 clinical trial is planned to be conducted in Canada and targeted to recruit up to 48 and 24 healthy volunteers for the single-ascending dose (SAD) and multiple- ascending dose (MAD) cohorts, respectively. This press release is provided “as is” without any representation or warranty of any kind.
The SCOPE (Summit for Clinical Ops Executives) conference is a premier event in the field of clinical operations, providing a comprehensive platform for professionals to explore the latest trends and innovations in clinical research. The conference will take place on February 11-14, 2024, in Orlando, Florida. Still not registered?
(NASDAQ: AXSM), a biopharmaceutical company developing novel therapies for the management of central nervous system (CNS) disorders, today announced positive results from the open-label Phase 2 COMET-TRD trial of AXS-05 in patients with treatment resistant depression (TRD). AXS-05 was well tolerated in the COMET trial.
Fast Track designation is well-timed, as we anticipate starting our Phase 2 clinical trial in hospitalized COVID-19 patients this month, and should help bring Brilacidin to patients faster in these dire times.”. Brilacidin, a versatile compound with broad therapeutic potential, is in a new chemical class called defensin-mimetics.
In the clinical research space, GxP is a set of quality regulations and guidelines designed to establish the safety, efficacy, and integrity of pharmaceuticals, medical devices, and clinical trials. This blog explores key concepts, regulations, and the importance of GxP in delivering successful clinical trials.
To protect human subjects in clinical research, the Food and Drug Administration (FDA) maintains and enforces specific regulations. Research stakeholders must track, document, and store the required information for trial oversight and monitoring to comply with regulations. What is an Electronic Master File (eTMF)? .
As the research community continues to seek clinical trial efficiencies designed to bring therapies to patients faster, more governing bodies are pushing for single institutional review board (single IRB) review as one way to streamline efforts. Policies and Regulations Affecting Single IRB Review. Common Rule Single IRB Policy.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.















































Let's personalize your content