This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Dive into this week’s update for more details on the actions taken by the FDA in the ongoing response to the Covid-19 pandemic. FDAapproves first treatment for Covid-19. On October 22, the FDAapproved the antiviral drug Veklury for use in adult and pediatric patients for the treatment of Covid-19 requiring hospitalization.
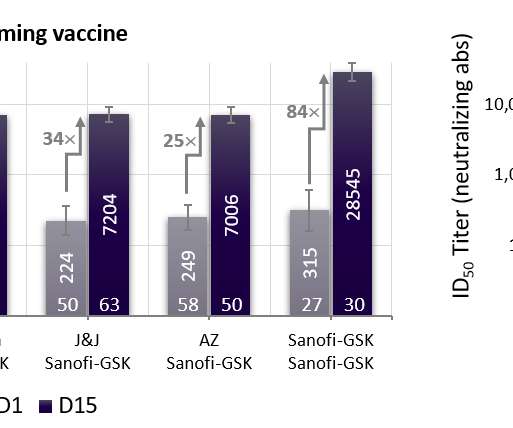
* Final analysis of the global VAT02 booster trial confirms universal ability to boost neutralizing antibodies 18- to 30-fold across vaccine platforms (mRNA, adenovirus). * efficacy against any symptomatic COVID-19 disease, in line with expected vaccine effectiveness in today’s environment dominated by variants of concern.
November 16, 2020 — An independent data and safety monitoring board (DSMB) overseeing the Phase 3 trial of the investigational COVID-19 vaccine known as mRNA-1273 reviewed trial data and shared its interim analysis with the trial oversight group on Nov. The interim analysis comprised 95 cases of symptomatic COVID-19 among volunteers.
Food and Drug Administration (FDA) to support the evaluation of a third, or booster, dose of the companies’ COVID-19 vaccine (BNT162b2) for future licensure. The data we’ve seen to date suggest a third dose of our vaccine elicits antibody levels that significantly exceed those seen after the two-dose primary schedule.
We believe that our investments in mRNA delivery technology and manufacturing process development will allow us to store and ship our COVID-19 vaccine candidate at temperatures commonly found in readily available pharmaceutical freezers and refrigerators,” said Juan Andres, Chief Technical Operations and Quality Officer at Moderna. “We
Vaccine candidate was found to be more than 90% effective in preventing COVID-19 in participants without evidence of prior SARS-CoV-2 infection in the first interim efficacy analysis.
This means that protection is achieved 28 days after the initiation of the vaccination, which consists of a 2-dose schedule.
Food and Drug Administration approved Pfizer’s coronavirus vaccine for emergency use on Friday, clearing the way for the launch of a national campaign to inoculate enough Americans to stem the spread of COVID-19. ” Who is first in line to be vaccinated? SATURDAY, Dec. 12, 2020 – The U.S.
First approval of a conjugate vaccine that helps protect against 20 serotypes responsible for the majority of invasive pneumococcal disease and pneumonia, 1,2,3,4,5,6,7 including seven responsible for 40% of pneumococcal disease cases and deaths in the U.S. Following today’s FDAapproval, the U.S. Jansen, Ph.D.,
Food and Drug Administration authorized a second booster dose of either the Pfizer-BioNTech or the Moderna COVID-19 vaccines for older people and certain immunocompromised individuals. The FDA previously authorized a single booster dose for certain immunocompromised individuals following completion of a three-dose primary vaccination series.
.
Pfizer-BioNTech files for US approval of COVID-19 vaccine ( Reuters ) ( NYTimes ) ( Politico ) ( Press ).
Covid-19 Vaccines Are Wasted as Special Syringes Run Short ( WSJ ).
Adcomm splits slightly in favor of FDAapproving ChemoCentryx’s rare disease drug ( Endpoints ).
Food and Drug Administration (FDA) has issued Emergency Use Authorization (EUA) for its single-dose COVID-19 vaccine, developed by the Janssen Pharmaceutical Companies of Johnson & Johnson, to prevent COVID-19 in individuals 18 years of age and older. The terms of the EUA allow use of the vaccine while more data are gathered.
Food and Drug Administration (FDA) advisory panel in regard to the COVID-19 vaccine from Moderna. The Vaccines and Related Biological Products Advisory Committee will be determining whether the product should be authorized for emergency use, according to CNBC. The vote itself is not slated to take place until after 3 p.m.
Food and Drug Administration issued an emergency use authorization (EUA) for the second vaccine for the prevention of coronavirus disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The emergency use authorization allows the Moderna COVID-19 Vaccine to be distributed in the U.S. Hahn , M.D.
Rare cases of the neurological disorder, Guillain-Barré syndrome have been reported following vaccination with the Janssen COVID-19 vaccine. Most occurred within 42 days after vaccination. For further information on the safety of authorized COVID-19 vaccines, please visit: [link]. What Is the Janssen COVID-19 Vaccine?
Food & Drug Administration (FDA) has extended the shelf life for the Johnson & Johnson single-shot COVID-19 vaccine to six months. Expiration dates will be updated on www.vaxcheck.jnj , where vaccine providers can confirm the latest expiration dates of our vaccine. We continue to work with the U.S.
Food and Drug Administration is expected to approve emergency use of Pfizer’s coronavirus vaccine as early as Saturday after its advisory panel cleared the way for the start of a national campaign to inoculate Americans and stem the spread of COVID-19. Who is first in line? Centers for Disease Control and Prevention.
Food and Drug Administration advisory panel will vote on Thursday whether to recommend emergency approval of Pfizer’s coronavirus vaccine, a decision that will come not a moment too soon as the country reported more than 3,000 new COVID-19 deaths on Wednesday. Meanwhile, U.S.
Doses are being supplied for use pursuant to the FDA Emergency Use Authorization for high-risk patients with mild to moderate COVID-19 in order to reduce the risk of progression to severe COVID-19 and/or hospitalization. Regeneron Pharmaceuticals, Inc. NASDAQ: REGN ) today announced that the U.S. Schleifer , M.D., ” The U.S.
(Nasdaq: MRNA), a biotechnology company pioneering messenger RNA (mRNA) therapeutics and vaccines to create a new generation of transformative medicines for patients, today announced that the primary efficacy analysis of the Phase 3 study of mRNA-1273 conducted on 196 cases confirms the high efficacy observed at the first interim analysis.
Food and Drug Administration (FDA) to begin a Phase I clinical trial of hAd5-COVID-19, the company’s novel COVID-19 vaccine candidate that targets both the inner nucleocapsid (N), engineered to activate T cells, and outer spike (S) protein, engineered to activate antibodies against the coronavirus (SARS-CoV-2).
January 29, 2021 – Johnson & Johnson (NYSE: JNJ) (the Company) today announced topline efficacy and safety data from the Phase 3 ENSEMBLE clinical trial, demonstrating that the investigational single-dose COVID-19 vaccine in development at its Janssen Pharmaceutical Companies met all primary and key secondary endpoints.
Casirivimab and imdevimab are not authorized for use in patients who are hospitalized or require oxygen therapy due to COVID-19, or for people currently using chronic oxygen therapy because of an underlying comorbidity who require an increase in baseline oxygen flow rate due to COVID-19. . “The Yancopoulos, M.D.,
Description: I’ve been a researcher for over 15 years at one of the biggest hospitals in Cleveland, Ohio. Every capsule is made here, in the USA, in our FDAapproved and GMP certified facility, under sterile, strict and precise standards. DermaPrime Plus capsules are non-GMO and safe.
Launch activities continue following the full FDAapproval for second-line metastatic triple-negative breast cancer (“mTNBC”) and accelerated approval for metastatic urothelial cancer. Sales of Veklury are generally affected by COVID-19 related rates of infections, hospitalizations and vaccinations. billion and $3.1
You are trying to lose weight, you are not sick, so why eat tasteless, hospital food? Every capsule is manufactured in the USA, in our FDAapproved and GMP certified facility, under sterile, strict and precise standards. So stick to plain water or unsweetened tea if you want to see some results.
Food and Drug Administration (FDA) is plenty busy with COVID-19 vaccine Emergency Use Authorizations (EUAs) this month, but they’re also wrapping up the year with a few PDUFA dates for other therapies. The FDAapproval of Klisyri is a significant milestone for Athenex,” said Johnson Lau, chairman and chief executive officer of Athenex.
Casirivimab and imdevimab are not authorized for use in patients who are hospitalized or require oxygen therapy due to COVID-19, or for people currently using chronic oxygen therapy because of an underlying comorbidity who require an increase in baseline oxygen flow rate due to COVID-19. . Yancopoulos , M.D.,
In a clinical trial of patients with COVID-19, casirivimab and imdevimab, administered together, were shown to reduce COVID-19-related hospitalization or emergency room visits in patients at high risk for disease progression within 28 days after treatment when compared to placebo. Hahn , M.D.
Another rejected proposal was a definition of “vaccine” (vaccines are exempt from Medicaid rebates), which would have limited this term to a product that is administered prophylactically – i.e., to prevent rather than treat a disease. 1396r-8(k)(3).
Food and Drug Administration (FDA) approval of Dupixent in September 2022 and the European Medicines Agency approval of Dupixent in December 2022. Dupixent is the only approved biologic indicated for prurigo nodularis in the U.S. Results from the trials were the basis for the U.S. Source link: [link]
This is a time when several promising treatments – including mRNA vaccines, BiTE therapies and CAR-T cell therapy, are essentially in competition with each other – they all have a common goal of treating the same disease, but they are approaching the objective from different angles.
During the call, Takeda provided a deep dive into TAK-721, which has the potential to be the first FDA-approved agent for the treatment of eosinophilic esophagitis (EoE), and TAK-003, which is a live attenuated tetravalent vaccine for prevention of dengue disease.
The top 10 news stories this week are focused on coronavirus vaccines and treatments, as China is set to move ahead with human testing of a potential vaccine that has been created using insect cells, while AstraZeneca has started a clinical trial of its antibody treatment designed to prevent and also treat symptoms of coronavirus.
hospitals are stretched to the limit trying to help people battling serious cases of COVID-19. This improvement stems from several factors, including the FDA’s emergency use authorization (EUA) of a number of therapies found to be safe and effective for COVID-19. Credit: NIH Right now, many U.S.
Wave 1 Q2 highlights include:
TAK-721 completed the rolling NDA submission and remains on track to be the first FDA-approved agent to treat eosinophilic esophagitis.
TAK-003 is on track for a regulatory filing for Dengue vaccine in endemic countries in Asia and Latin America, and in the EU in Q4 FY2020.
FDA’s guidance on developing products to prevent or treat Covid-19 Of the five guidance documents that received an extension, one addresses the development of drugs and biological products for Covid-19. It also stated that companies should consider whether an investigational product might interact with an administered vaccine for Covid-19.
FDA Actions. FDAApproval: Last week the FDAapproved Veklury (remdesivir) for the treatment of COVID-19 requiring hospitalization in adults and pediatric patients (12 years of age and older). Testing Therapies, Antivirals and Vaccines. Please read more here. . Days after the U.S.
at CER driven by growth drivers Dupixent ® and Vaccines. Vaccines up 5.3%, driven by PPH franchise and demand for influenza vaccines in southern hemisphere. Vaccines delivered growth in its core segments. Polio/Pertussis/Hib vaccines (incl. Influenza vaccines (incl. Meningitis/Pneumo vaccines (incl.
Despite the initial promise of PFCs and FDAapproval of a product called Fluosol-DA in 1989, PFCs have many drawbacks. Hemopure, the only FDA-approved HBOC product, is a bovine hemoglobin that is chemically crosslinked (that is, several hemoglobin molecules are bound to each other) to improve its stability.
FDAApproves Verquvo (vericiguat) for Heart Failure with Reduced Ejection Fraction. Patients with symptomatic chronic heart failure and reduced ejection fraction have a high risk for hospitalization after experiencing symptoms of heart failure requiring outpatient IV diuretic treatment or hospitalization. KENILWORTH, N.J.–(BUSINESS
“The death toll from COVID-19 continues to rise around the world and hospitalizations, particularly in the U.S., These data further support our belief that bamlanivimab and etesevimab together have the potential to be an important treatment that significantly reduces hospitalizations and death in high-risk COVID-19 patients.
Direct Relief will allocate donations of baricitinib to low- and lower-middle-income countries (based on World Bank classification) for the treatment of hospitalized COVID-19 patients requiring supplemental oxygen, based on requests from these governments to Direct Relief. Ricks , Lilly chairman and CEO.
In this special report, I’m going to show you why researchers at Harvard University and the Massachusetts General Hospital in Boston are now praising a strange new pain treatment originally discovered on the International Space Station. It is FDA-registered and manufactured in the USA at an FDAapproved plant. Absolutely!
It also defines LBPs as “A biological product that: 1) contains live organisms, such as bacteria; 2) is applicable to the prevention, treatment, or cure of a disease or condition of human beings; and 3) is not a vaccine.” AgencyIQ is planning a deeper dive into these recent actions by the FDA. What’s next?
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.









































Let's personalize your content