This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
FDAApproves Sesquient (fosphenytoin sodium) for the Treatment of Status Epilepticus in Adult and Pediatric Patients. Food and Drug Administration (FDA) has approved Sesquient (fosphenytoin sodium for injection) for the treatment of status epilepticus in adult and pediatric patients. About Sesquient.
NYSE: PFE) and BioNTech SE (Nasdaq: BNTX) today announced they have submitted a supplemental Biologics License Application (sBLA) to the U.S. Food and Drug Administration (FDA) to expand the approval of COMIRNATY® (COVID-19 Vaccine, mRNA) to include individuals ages 12 through 15 years. Pfizer Inc.
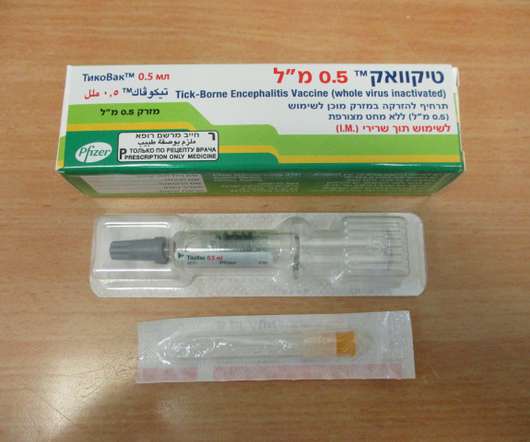
Food and Drug Administration (FDA) has approved TICOVAC (tick-borne encephalitis (TBE) vaccine) for active immunization to prevent TBE in individuals 1 year of age and older. 1 TICOVAC is the only FDA-approved vaccine to help protect U.S. Following today’s FDAapproval, the U.S. Source link: [link].
FDAApproves Danyelza (naxitamab-gqgk) for the Treatment of Neuroblastoma. It’s very exciting to see this treatment go from being an experimental therapy used at my daughter’s bedside to now being FDAapproved. See below for information related to adverse reactions. Important Safety Information. Indication.
Valentine — On November 22, 2022, FDAapproved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
Food and Drug Administration (FDA) approval of Darzalex Faspro ® (daratumumab and hyaluronidase-fihj), a subcutaneous formulation of daratumumab, in combination with bortezomib, cyclophosphamide and dexamethasone (D-VCd) for the treatment of adult patients with newly diagnosed light chain (AL) amyloidosis.[1]
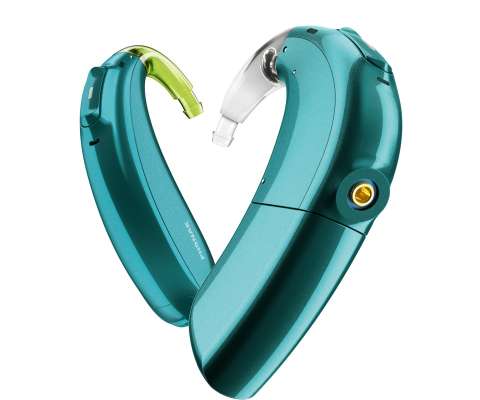
.–( BUSINESS WIRE )– Advanced Bionics (AB) , a global leader in cochlear implant technology, in collaboration with Phonak, a leading provider of life-changing hearing solutions, receives FDAapproval and announces it is bringing Marvel hearing technology to Advanced Bionics cochlear implant wearers. About Advanced Bionics.
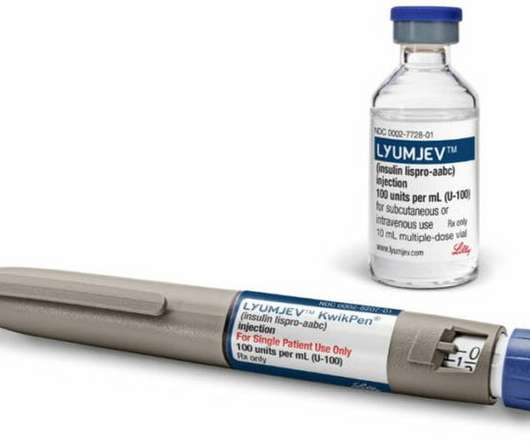
For more information, call 1-800-545-5979 or go to www.Lyumjev.com or www.humalog.com. This summary provides basic information about Lyumjev and Humalog. It does not include all information known about these medicines. Read the information that comes with your prescription each time your prescription is filled.
FDA unveils long-awaited Patient Medication Information proposed rule Since 2017, the FDA has been working on a proposal to create a new type of patient-focused labeling for certain outpatient drug products that would be specifically targeted for patient use.
CNS holds a worldwide exclusive license to the Berubicin chemical compound and has acquired all data and know-how from Reata Pharmaceuticals, Inc.
For more information, please visit www.CNSPharma.com.
Forward-Looking Statements .
WAKIX is the first and only treatment approved by the FDA for people with excessive daytime sleepiness or cataplexy associated with narcolepsy that is not scheduled as a controlled substance by the U.S. WAKIX received FDAapproval for the treatment of excessive daytime sleepiness in adult patients with narcolepsy in August 2019.
1, 2020 /PRNewswire/ — Sosei Group Corporation (“the Company”) (TSE: 4565) announces it has entered into a global collaboration and license agreement with Biohaven Pharmaceutical Holding Company Ltd. (“Biohaven”, NYSE: BHVN). For more information, please visit [link]. About Biohaven. About CGRP.
Lilly is offering donations of baricitinib to the Indian government through Direct Relief while simultaneously working with local Indian pharmaceutical companies to execute royalty-free voluntary licensing agreements to accelerate the manufacturing and distribution of the medicine in India during the pandemic.
FDA 12/1/2022, To treat adults with relapsed or refractory acute myeloid leukemia with a susceptible isocitrate dehydrogenase-1 (IDH1) mutation, Rezlidhia Olutasidenib , sold under the brand name Rezlidhia , is an anticancer medication used to treat relapsed or refractory acute myeloid leukemia. [1] 1] It is taken by mouth. [1] Hz, 1 H), 4.62−4.75
News information is not all-inclusive and updates are published once a week on Tuesdays. . . FDA Actions. FDAApproval: Last week the FDAapproved Veklury (remdesivir) for the treatment of COVID-19 requiring hospitalization in adults and pediatric patients (12 years of age and older). Other Industry News.
Second FDAapproved indication for dostarlimab in 2021 GARNET study demonstrated objective response rate of 41.6% This indication received accelerated approval based on tumour response rate and durability of response. This approval was based on data from cohort A1, which included 71 patients with dMMR endometrial cancer.
“In the interest of public health and safety, it remains a priority to provide healthcare professionals with as much information as possible about treatment options that may help improve outcomes for patients with severe disease,” said Ilya Yuffa, senior vice president and president of Lilly Bio-Medicines.
2 , 3 Lazertinib was first approved in South Korea on January 18, 2021, for the treatment of EGFR T790M mutation-positive non-small cell lung cancer (NSCLC) with EGFR mutations. 1 It was approved by the FDA on August 19, 2024. 1 It was approved by the FDA on August 19, 2024. Food and Drug Administration (FDA).
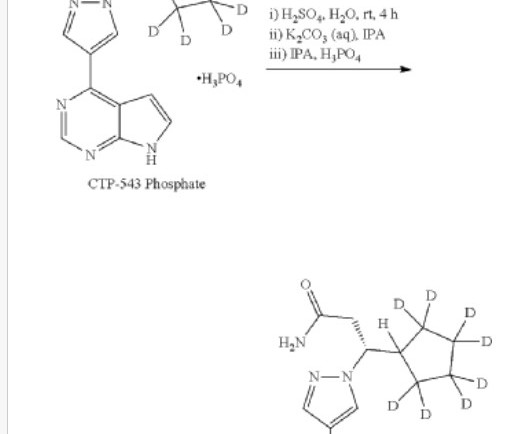
Deuruxolitinib C 17 H 18 N 6 , 314.422 Fdaapproved Leqselvi , 7/25/2024, To treat severe alopecia areata C-21543, CTP 543, CTP-543, CTP543 (3r)-3-(2,2,3,3,4,4,5,5-d8)cyclopentyl-3-(4-(7h-pyrrolo(2,3-d)pyrimidin-4-yl)-1h-pyrazol-1-yl)propanenitrile 1h-pyrazole-1-propanenitrile,beta.-(cyclopentyl-2,2,3,3,4,4,5,5-d8)-4-(7h-pyrrolo(2,3-d)pyrimidin-4-yl)-,
If approved, Actemra/RoActemra would be the first U.S. FDAapproval is expected in the second half of this year. Food and Drug Administration (FDA)-approved for this use and there is limited information known about the safety or effectiveness of using Actemra/RoActemra to treat people in the hospital with COVID-19.
While the chance of having this occur is very low, Johnson & Johnson has updated its COVID-19 Vaccine Factsheet to include important information about these rare cases and on the signs and symptoms of Guillain-Barré syndrome. For further information on the safety of authorized COVID-19 vaccines, please visit: [link].
1] Motixafortide was approved for medical use in the United States in September 2023. [2] 4 Similar in mechanism to the previously approved plerixafor , motixafortide is an inhibitor of C-X-C Motif Chemokine Receptor 4 (CXCR4), a protein that helps to anchor stem cells to bone marrow matrix. 1] It is given by subcutaneous injection. [1]
Food and Drug Administration (FDA) on Nov. Please see below for important warnings and information about the authorized use of baricitinib in the U.S. Baricitinib, an oral JAK inhibitor, was discovered by Incyte and licensed to Lilly. It is approved and commercially available as OLUMIANT in the U.S. Serious Infections: ?Serious
S2CID 250989659. ^ “Eplontersen: FDA-Approved Drugs” U.S. Food and Drug Administration (FDA). Retrieved 21 December 2023. ^ “Wainua (eplontersen) granted regulatory approval in the U.S. 88 (12): 5389–5398. doi : 10.1111/bcp.15468. PMID 35869634. Retrieved 22 December 2023. hdl : 10665/340684.
For information on the authorized use of baricitinib and mandatory requirements under the EUA, please review the FDA Letter of Authorization , Fact Sheet for Healthcare Providers and Fact Sheet for Patients, Parents and Caregivers ( English ; Spanish ). It is approved in the U.S.
Veklury is contraindicated in patients who are allergic to Veklury or any of its components; please see below for additional Important Safety Information for Veklury. Veklury is contraindicated in patients who are allergic to Veklury or any of its components; please see below for additional Important Safety Information for Veklury.
Molecular Weight: 631.700 FDAAPPROVED, To treat moderately to severely active ulcerative colitis in adults, 10/12/2023 Velsipity Etrasimod , sold under the brand name Velsipity , is a medication that is used for the treatment of ulcerative colitis (UC). [1] “FDAApproves New Drug for Ulcerative Colitis” Medscape.
For instance, it would be acceptable for a biosimilar’s labeling to differ from its reference product for the purpose of providing information necessary to inform its safe and effective use (e.g., In 2018, the FDA published a detailed guidance on labeling requirements for biosimilar products. Yes, the guidance confirms.
The Company plans to file for a Biologics License Application (BLA) with the FDA later in 2021. Manufacturing and Supply Chain Information. The Janssen COVID-19 vaccine has not been approved or licensed by the U.S. There is no FDA-approved vaccine to prevent COVID-19. Important Safety Information.
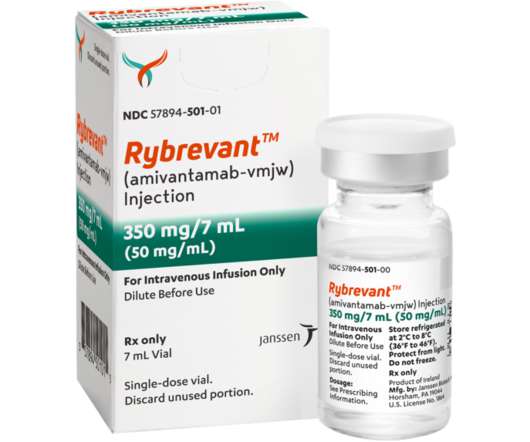
Food and Drug Administration (FDA) approval for patients with locally advanced or metastatic NSCLC with epidermal growth factor receptor (EGFR) exon 20 insertion mutations, based on data showing an ORR of 40 percent (95 percent CI, 29 – 51) and median duration of response of 11.1 RYBREVANT™ IMPORTANT SAFETY INFORMATION 7.
All products are received in one of two forms: A New Drug Application (or NDA, for pharmaceuticals) or a Biologics License Application (BLA, for biologics). Novel drug products are defined as products that have never been approved for any indication. Data on these novel approvals is published throughout the year by both CDER and CBER.
There are currently no FDA-approved treatments for AA. ” Baricitinib is an oral JAK inhibitor discovered by Incyte and licensed to Lilly. It is approved and commercially available as OLUMIANT in the U.S. IMPORTANT SAFETY INFORMATION FOR OLUMIANT (baricitinib) TABLETS . USE IN SPECIFIC POPULATIONS.
Food and Drug Administration (FDA) has extended the Prescription Drug User Fee Act (PDUFA) date for review of the Company’s Biologics License Application (BLA) seeking accelerated approval of pegunigalsidase alfa (PRX–102) for the proposed treatment of adult patients with Fabry disease. Protalix has licensed to Pfizer Inc.
Under the terms of the settlement agreement, the litigation between the parties in the United States District Court for the District of New Jersey will be ended, and Lupin will have a license to sell its generic product beginning April 2033, or earlier under certain circumstances. IMPORTANT SAFETY INFORMATION.
The application will be reviewed by the EMA’s Committee for Medicinal Products for Human Use (CHMP) under the centralized licensing procedure for all 27 Member States of the European Union, as well as Norway, Iceland and Liechtenstein. For further information, please see [link]. About Lenacapavir.
Brilacidin for UP/UPS was licensed to Alfasigma S.p.A. A Phase 2b trial of Brilacidin showed a single intravenous dose of the drug delivered comparable outcomes to a seven-day dosing regimen of the FDA-approved blockbuster daptomycin in treating Acute Bacterial Skin and Skin Structure Infection.
As FDA explains, Boehringer’s request would allow its “Cyltezo (adalimumab-adbm) injection, which contains the same total content of drug substance and same concentration as Original Concentration Humira (e.g., mL), to be biosimilar to or interchangeable with High Concentration Humira (e.g., mL) in addition to Original Concentration Humira.”
Food and Drug Administration (FDA) has extended the review period for the supplemental New Drug Application (sNDA) for baricitinib for the treatment of adults with moderate to severe atopic dermatitis (AD). OLUMIANT is a once-daily, oral JAK inhibitor approved in the U.S. See the full Prescribing Information here.
The FDA grants Emergency Use Authorization to medicines that may help diagnose, treat or prevent a life-threatening disease when adequate and approved alternatives are not available. The EUA is temporary and does not take the place of a formal biologics license application (BLA) submission review and approval process.
–( BUSINESS WIRE )– Bristol Myers Squibb (NYSE: BMY) today announced that the Biologics License Application (BLA) for lisocabtagene maraleucel (liso-cel) for the treatment of adults with relapsed or refractory (R/R) large B-cell lymphoma after at least two prior therapies remains under review by the U.S. 1, 2021 11:59 UTC.
if approved Acceptance marks another important step in advancing Dupixent for a broad range of diseases with underlying type 2 inflammation. The target action date for the FDA decision is September 30, 2022. For additional information about the company, please visit www.regeneron.com or follow @Regeneron on Twitter.
FDA-approved labeling for OLUMIANT includes a Boxed Warning for Serious Infections, Malice, and Thrombosis. See the full Prescribing Information then. Marketing authorization for the treatment of rehabilitated cases with COVID-19 has been granted for OLUMIANT in multiple countries.
“Alopecia areata is a challenging disease that currently has no FDA-approved treatment options, making it difficult for healthcare providers to best serve the needs of these patients,” said Brett King , MD, PhD associate professor of Dermatology at Yale School of Medicine.
The intended benefit for sponsors of this approach is that products making use of designated platforms would be eligible to receive expedited development benefits from the FDA, including access to early-stage development meetings with regulators. However, both deadlines came and went without any official action by the FDA.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.









































Let's personalize your content