This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
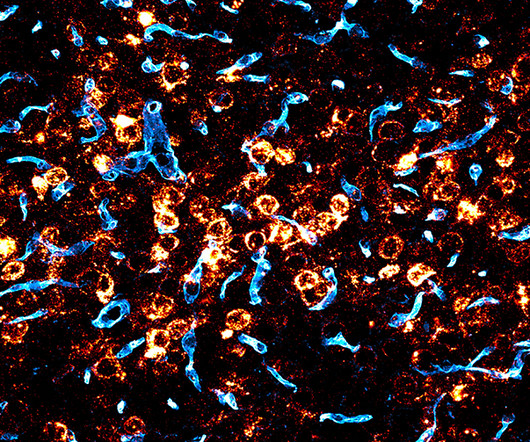
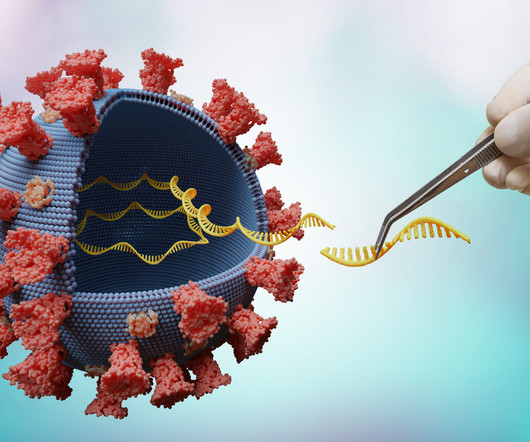
But FDA-approved forms of the most commonly used vehicle for packaging and delivering these therapies to target cells, adeno-associated viruses (AAVs), aren’t able to efficiently cross the blood-brain barrier at high levels and deliver therapeutic cargo.
Valentine — On November 22, 2022, FDAapproved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
A significant effort has been made to correctly map the drug form of the EMA data by manually inspecting different EMA sources of information, such as the Product Information (Annex I: Summary of Product Characteristics and Annex III: Labelling and Package Leaflet) and/or Assessment Report, where available.
Biotechnology services include advising or consulting services related to the use of the above equipment, disease detection, or genealogical information, or any other service for the research, development production, analysis, detection, or provision of information including data storage and transmission related to biological materials.
FDA unveils long-awaited Patient Medication Information proposed rule Since 2017, the FDA has been working on a proposal to create a new type of patient-focused labeling for certain outpatient drug products that would be specifically targeted for patient use.
When returned directly to participants, aggregate results should be packaged and communicated in user-friendly formats, such as in newsletters or web pages constructed with readability and health literacy principles in mind. Conversely, a CGM not yet FDA-approved assessed in the study for reliability, would yield an investigational result.
The authority to change drug labels outside of considerations for new safety information “could encourage third parties, such as academic investigators, insurance companies, and cooperative trial groups, to initiate such changes,” they wrote. .
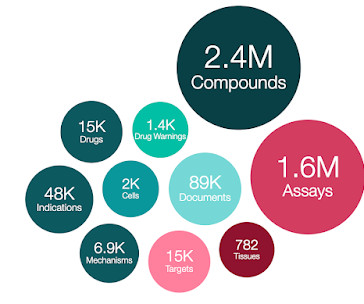
This field was populated with mode of action information that had previously been recorded as metadata in the ASSAY_PARAMETERS and ACTIVITY_PROPERTIES tables. Data changes and amendments Missing information in the field DOCS.YEAR was included for 385 patents (src_id = 38). In addition, approx. 1 = yes, 0 = default value).
End-to-End Single-Site Solution from Drug Substances to Fill-Finish & Packaging. In addition to drug substance manufacture, the facility will also provide commercial scale, automated fill-finish and assembly, packaging and labeling services. Packaging line?.
For more information, please visit: www.fujifilmholdings.com.
It is critical that the nonclinical program outlined in the PIND briefing document is presented in a manner that allows FDA to provide relevant input on the required IND-enabling studies. For example, if the nonclinical data presented in the PIND briefing document lacks information on the potential toxicity profile of the drug (e.g.,
The product is made here, in the USA, in our FDAapproved and GMP certified facility, under sterile, strict and precise standards. After you fill in your information and confirm your package will be shipped for free right to your doorstep as soon as possible. Dentitox Pro is non-GMO and safe.
It is worth noting that this is not the same as a full approval, which requires additional information. To this point, Moderna has only submitted two months of follow-up safety data, and the FDA typically requires six months for a full approval. Initial doses are expected to be limited as manufacturing ramps up.
Unlike the traditional 505(b)(1) pathway, which requires extensive original investigations, the 505(b)(2) pathway allows developers to rely on information contained in published literature and/or from studies not conducted by the applicant, which can significantly reduce development time and cost. daily versus four times daily dosing).
For more information on key components of a fit-for-purpose, phase-by-phase QMS guidance download Advarra’s whitepaper Steps to Implementing a Quality Management System. The auditor prepares by reviewing relevant information, such as contracts, as well as select QMS documents and records. Q: What must be done to prove oversight?
Food and Drug Administration – labeled an “Official Action Indicated” classification – is generally devastating for the facility, not least because it can stall FDAapproval of applications to market drugs manufactured at the facility. Farquhar — A drug manufacturer’s bad post-inspection grade from the U.S.
CMS did not finalize the price verification survey, which would have required manufacturers of 10 costly drugs selected annually to provide clinical information as well as information on production, distribution, research, and marketing costs, revenue and profit, and ex-U.S. pricing, among other things.
The treatment had been granted breakthrough therapy designation, but FDAapproval would ultimately rest on the sponsor’s ability to demonstrate clinically meaningful improvement. The FDA also granted a second meeting for review. The agency encouraged further review of the data for potential reference as a historical control.
Every capsule is made here, in the USA, in our FDAapproved and GMP certified facility, under sterile, strict and precise standards. After you fill in your information and confirm your order, your bottle of DermaPrime Plus will be shipped for free right to your doorstep as soon as possible.
Prior to CARES Act reforms, FDA relied on a monograph process through which firms could bring OTC drugs to market without FDAapproval so long as they adhered to pre-set terms under the monograph. On the other hand, data, information and comments to a proposed or final order should use the OTC Monographs@FDA portal.
While the definition of NME has changed over the years, it can sometimes include a combination product consisting of at least one drug that has previously been approved. Data on these novel approvals is published throughout the year by both CDER and CBER. FDAapproved 13 NMEs through the AA pathway in FY 2023, making up 25.5%
The difference at issue here is the fact that Vanda’s Hetlioz product includes braille writing on its packaging, with some accompanying language (“Do not cover Braille”), and the generic products’ labeling does not. Vanda’s Hetlioz was, in fact, the first FDA-approved drug product to include braille labeling. . § 314.94(a)(8)(iv),
. | NOV 8, 2023 10:02 PM CST Regulatory background The Federal Food, Drug, and Cosmetics Act (FD&C Act) is administered by the Food and Drug Administration (FDA) and regulates cosmetics. with a contact agent for the facility, as well as electronic contact information (if available). Foreign facilities will provide the U.S.
See “Worldwide Pro Forma Revenue” in Quarterly Package of Financial Information for this quarter, which is available on bms.com/investors/financial-reporting/quarterly-results, for information on the revenue of the company and Celgene on a stand-alone basis for the prior-year period. Otezla ® is a trademark of Amgen Inc.
After all my requirements were 100% met, we finally had the final product: Every capsule is made here, in the USA, in our FDAapproved and GMP certified facility, under sterile, strict and precise standards.Met Slim Pro capsules are non-GMO and safe. Safe: processed under strict sterile standards with regularly disinfected equipment.
Once samples are validated, this will conclude the IND package” said Kyle Klepner, Senior Scientist, Study Director, Safety Assessment, at Altasciences. Virpax is initially seeking FDAapproval for two prescription drug candidates that employ two different patented drug delivery platforms.
Every capsule is manufactured in the USA, in our FDAapproved and GMP certified facility, under sterile, strict and precise standards. After you fill in your information and confirm your order, your bottle of Alpha Zym Plus will be shipped for free right to your doorstep as soon as possible.
Health care providers should review the Fact Sheet for detailed information on the authorized use and requirements of the EUA and may call 844-734-6643 for more information. Please see the Fact Sheet and FDA Letter of Authorization at [link]. AUTHORIZED USE AND IMPORTANT SAFETY INFORMATION. Authorized Emergency Use.
Regeneron is also informing the U.S. Founded and led for over 30 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to eight FDA-approved treatments and numerous product candidates in development, all of which were homegrown in our laboratories.
1 In 2017, the US Food and Drug Administration (FDA) approved the first AAV-based gene replacement therapy (Luxturna), for Leber congenital amaurosis type 2. 2 Since then, the FDA has approved four more AAV-based gene therapies—Zolgensma, Hemgenix, Elevidys and Rocktavian—for treating various diseases.
As healthcare professionals, it’s our responsibility to educate patients about generic drugs and empower them to make informed decisions about their treatment options. Explain the FDAApproval Process Many patients are unaware of the rigorous approval process generic drugs must undergo.
AUTHORIZED USE AND IMPORTANT SAFETY INFORMATION. Authorized Emergency Use Casirivimab and imdevimab injection is an investigational combination therapy and has been authorized by FDA for the emergency use described above. Casirivimab and imdevimab injection is not FDAapproved for any use.
IMBRUVICA is the only FDA-approved medicine in WM and cGVHD. IMBRUVICA was one of the first medicines to receive FDAapproval via the Breakthrough Therapy Designation pathway. For more information, visit www.IMBRUVICA.com. Venetoclax is approved in more than 80 countries, including the U.S. Information.
AUTHORIZED USE AND IMPORTANT SAFETY INFORMATION. Casirivimab and imdevimab injection is an investigational combination therapy and has been authorized by FDA for the emergency use described above. Casirivimab and imdevimab injection is not FDAapproved for any use. Authorized Emergency Use.
sensitivity and specificity) – including colonoscopy procedures and an FDA-approved stool-based test ( Cologuard ) – but comparatively low uptake, given the prevalence of CRC in the population. Notably, CRC screening methods are currently available and have generally high performance (i.e.,
Such a situation is commonplace in the clinical trial realm, in which investigational drug products which are not already FDAapproved are administered to patients. Under the Pandemic and All-Hazards Preparedness Reauthorization Act of 2013 (PAHPRA) , the FDA also has some authority to extend MCM expiration dates.
FDAapproval for adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response or intolerance to methotrexate. Use and Important Safety Information 14. What is the most important information I should know about RINVOQ? For more information, talk to your HCP.
AUTHORIZED USE AND IMPORTANT SAFETY INFORMATION Authorized Emergency Use Casirivimab and imdevimab injection is an investigational combination therapy and has been authorized by FDA for the emergency use described above. Casirivimab and imdevimab injection is not FDAapproved for any use.
FDA now intends to release that regulation in August 2023 – just two months from now, indicating that this effort was proceeding apace behind-the-scenes for some time since these types of regulations often take years to develop.
“FDA requested additional data to support effectiveness, such as evidence showing that plasma arginine and metabolite reduction predicts a clinical benefit in patients.” Additionally, the agency requested further information regarding chemistry, manufacturing and controls (CMC). The clinical package, safety and label were not affected.
Founded and led for 30 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to eight FDA-approved treatments and numerous product candidates in development, all of which were homegrown in our laboratories.
IMBRUVICA is the only FDA-approved medicine in WM and cGVHD. IMBRUVICA was one of the first medicines to receive FDAapproval via the Breakthrough Therapy Designation pathway. For more information, visit www.IMBRUVICA.com . Important Side Effect Information. Information. Do not use IMBRUVICA ® ?for
Health care providers should review the Fact Sheet for detailed information on the authorized use and requirements of the EUA and may call 844-734-6643 for more information. Please see the Fact Sheet and FDA Letter of Authorization at [link]. AUTHORIZED USE AND IMPORTANT SAFETY INFORMATION. Authorized Emergency Use.
FDA accepted for priority review Libtayo ® (cemiplimab-rwlc) for both advanced non-small cell lung cancer and basal cell carcinoma. FDAapproved Inmazeb for Ebola ( Zaire ebolavirus). See further details in the “ Other Financial Information ” section below. See note (4) below for further information.
Founded and led for over 30 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to eight FDA-approved treatments and numerous product candidates in development, all of which were homegrown in our laboratories.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.















































Let's personalize your content