This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Importantly, the 2018 Farm Bill preserved FDA authority to regulate products with cannabis or cannabis-derived compounds under the Federal Food, Drug, and Cosmetic (FD&C) Act and Section 351 of the Public Health Service Act. The FDA is granting INDs for such CBD research. Scientific and Ethical Challenges.
Karst — Listing patent information in the Orange Book is a matter of judgment, but that judgment call is about to get a bit more scrutiny. The bill proposes to amend FDC Act § 505(b) and PHS Act § 351(a)(2) with respect to patent information submitted to FDA for Orange Book and Purple Book listing. Koblitz & Kurt R.
Real-time prescription benefit tools are software applications that analyze a patient’s health insurance coverage and prescription requirements “by linking electronic health records with patients’ prescription benefits information.” The authors cite the FDA’s personal importation policy. Authors: Hussain S. Hwang, MD, MSPH; Aaron S.
Learn more about FDA-approved and authorized COVID-19 vaccines. Given the similar course of COVID-19 disease in adults and pediatric patients, today’s approval of Veklury in certain pediatric patients is supported by efficacy results from phase 3 clinical trials in adults. The FDA granted approval to Gilead Sciences Inc.
FDAApproves Danyelza (naxitamab-gqgk) for the Treatment of Neuroblastoma. This indication is approved under accelerated approvalregulation based on overall response rate and duration of response. See below for information related to adverse reactions. See below for information related to adverse reactions.
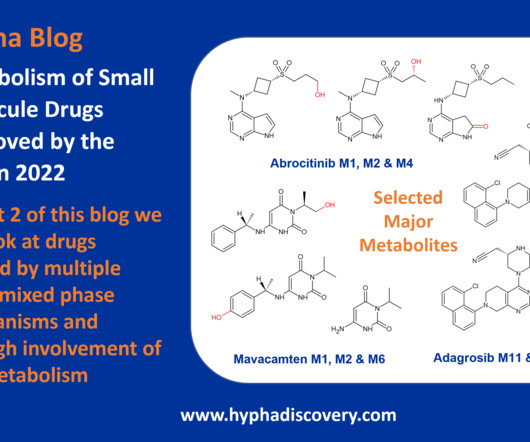
Metabolism of 2022 FDAapproved small molecule drugs part 2 Mixing it Up By Julia Shanu-Wilson In Part 1 of this topic we looked at metabolism of the small molecule drugs approved by the FDA in 2022 that were mediated by CYP3A4. Dermavant’s tapinarof is one such friend. 8 This is not the only point of interest.
The FDA previously granted Cytalux orphan-drug , priority and fast track designations. The FDA granted the approval to On Target Laboratories, LLC. Related Information. National Cancer Institute: Ovarian, Fallopian Tube, and Primary Peritoneal Cancer FDAApproved Drugs: Questions and Answers. ###.
People with achondroplasia have a genetic mutation that causes a certain growth regulation gene called fibroblast growth factor receptor 3 to be overly active, which prevents normal bone growth. A condition of this accelerated approval is a post-marketing study that will assess final adult height. Related Information.
Valentine — On November 22, 2022, FDAapproved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
FDAApproves Imcivree (setmelanotide) for Chronic Weight Management in Patients with Obesity Due to POMC, PCSK1 or LEPR Deficiency. With this approval, Imcivree becomes the first-ever FDAapproved therapy for these rare genetic diseases of obesity. Important Safety Information. BOSTON, Nov.
Livornese — I saw the sign…and the answer is no—FDA-approved labeling apparently is not enough under state failure-to-warn laws, according to certain courts. The GAO Report further explained that the agency did not have the resources to regulate the estimated 100,000 OTC drugs marketed through the monograph process.
Bile acids are crucial signalling molecules that aid the regulation of the immune system and have essential metabolic functions, like controlling lipid and glucose metabolism. microbeMAAST: a taxonomically informed mass spectrometry search tool for microbial metabolomics data. Biochemical Pharmacology. Nature Microbiology.
Food and Drug Administration (FDA) has approved commercial production at the company’s new CAR T-cell therapy manufacturing facility in Frederick, Maryland. The site will produce Kite’s FDAapproved CAR T-cell therapy used to treat blood cancer. For more information on Kite, please visit www.kitepharma.com.
Food and Drug Administration (FDA) approval of Darzalex Faspro ® (daratumumab and hyaluronidase-fihj), a subcutaneous formulation of daratumumab, in combination with bortezomib, cyclophosphamide and dexamethasone (D-VCd) for the treatment of adult patients with newly diagnosed light chain (AL) amyloidosis.[1]
See the prescribing information for additional information on risks associated with Jardiance. The FDA granted the approval of Jardiance to Boehringer Ingelheim. Related Information. FDA takes steps to spark development of heart failure drugs Heart Health for Women. ###. Source link: [link].
Treatment is First FDA-Approved Option Patients Can Take Regardless of Previous Therapies. Food and Drug Administration approved Besremi (ropeginterferon alfa-2b-njft) injection to treat adults with polycythemia vera, a blood disease that causes the overproduction of red blood cells. Related Information. Today, the U.S.
To further facilitate generic drug development, and to assist the generic pharmaceutical industry in this process, the FDA publishes product-specific guidances (PSGs) describing the agency’s current thinking and expectations on how to develop generic drug products that are therapeutically equivalent to their brand name counterparts.
FDA unveils long-awaited Patient Medication Information proposed rule Since 2017, the FDA has been working on a proposal to create a new type of patient-focused labeling for certain outpatient drug products that would be specifically targeted for patient use. The FDA recently concluded its work on a proposed rule focused on PMI.
On July 30, brand name drug sponsor Novartis asked the District Court for a Temporary Restraining Order ( here and here ) enjoining FDA’sapproval of a generic version of its ENTRESTO (sacubitril and valsartan) with certain dosing and indication information carved-out or modified.
The Act is intended to address national security concerns by prohibiting certain conduct by regulated industry. Effect on FDAapprovals— In cases where a biological product or drug needs to change aspects of its manufacturing processes to avoid using a covered equipment or service, will it need to file supplements with FDA for the CMC update?
Food and Drug Administration (FDA) approved a first-of-its-kind intentional genomic alteration (IGA) in a line of domestic pigs, referred to as GalSafe pigs, which may be used for food or human therapeutics.
The FDA granted approval of the IGA in GalSafe pigs to Revivicor Inc.
SILVER SPRING, Md. ,
The 51 regulations that FDA is currently working on The FDA today unveiled its much-anticipated Spring 2023 Unified Agenda, a document outlining the regulations the agency plans to release in 2023 and beyond. The anticipated date of publication is June 2023, meaning we should see this regulation imminently.
WAKIX is the first and only treatment approved by the FDA for people with excessive daytime sleepiness or cataplexy associated with narcolepsy that is not scheduled as a controlled substance by the U.S. WAKIX received FDAapproval for the treatment of excessive daytime sleepiness in adult patients with narcolepsy in August 2019.
.
In a Viewpoint, Bishal Gyawali, MD, PhD, and colleagues at the Program on Regulation, Therapeutics and Law (PORTAL) research group at Harvard Medical School and Brigham & Women’s Hospital suggested improving FDA’s authority to modify drug package insert for situations such as drug repurposing and de-escalation of therapy.
Gaulkin — In May 2023, we posted about a CMS proposed regulation that sought to make a wide variety of changes to the Medicaid Drug Rebate Program (MDRP), including a new “price verification survey,” and a controversial proposal to require “stacking” of discounts to different customers when determining best price.
A Wave of Innovations The year 2024 has already proven to be significant for the field of biologics, witnessing a notable increase in FDAapprovals. In the first quarter alone, the FDAapproved 9 new biologics, a substantial rise from the 5 approvals during the same period in 2023.
Specifically, Vanda alleges that FDA communications to certain generic competitors regarding dissolution rates, impurities, and micronization revealed Vanda’s confidential manufacturing information and caused economic injury to the company. FOOD AND DRUG ADMINISTRATION FOIA – FDA Presentation Pending 1:2023cv02325 (D.D.C.)
Under section §812.10, a sponsor may request a waiver of any requirement of the IDE regulations through an application with supporting documentation. The FDA may grant a waiver if the requirement is not stipulated by the Federal Food, Drug, and Cosmetic (FD&C Act) or if it is not necessary for the protection of human subjects. [3]
The agency will continue to evaluate data and information as it becomes available when considering the potential use of a second booster dose in other age groups. The FDA-authorized Pfizer-BioNTech COVID-19 Vaccine and the FDA-approved Comirnaty can be used to provide the authorized booster dose(s). Related Information.
156, a patent may be extended only once (even if it would be eligible for extension on more than one occasion because it applies to several FDA-approved products), and only one patent may be extended for each regulatory review period. In several letters to applicants, each styled as a REQUIREMENT FOR INFORMATION PURSUANT TO 37 C.F.R.
Food and Drug Administration (FDA)-approved therapies for treating PAH were primarily vasodilators, designed to overcome the imbalance between vasoactive and vasodilator mediators and to restore endothelial cell function. Historically, the available drugs and U.S.
This major shift to the orthodox tradition of using animal experiments in drug testing dates back the Aristotle’s time and cemented 80 years ago with initial federal mandate of drug safety regulation of 1938. To this end, the FDA’s newly created iSTAND initiative drives the path toward regulatory approval for devices like organ-on-chips.
The FDA intends to make background materials available to the public, including the meeting agenda and committee roster, no later than two business days prior to the meeting. 23, the FDA intends to issue a Federal Register notice with details of the meeting, which will include information about a public docket for comments.
As with other FDA-regulated products, such as human drugs and medical devices, the “regulatory review period” is composed of a “testing phase” and a “review phase.” The “review phase” is the period between the initial submission and approval of the NADA. FDA’s PTE regulations at 21 C.F.R.
Learn more Drug Repurposing Hub A curated and annotated collection of FDA-approved drugs, clinical trial drugs, and pre-clinical tool compounds. Learn more Genotype-Tissue Expression (GTEx) A comprehensive public resource to study tissue-specific gene expression and regulation.
Offshoring, sophisticated information technology, climate change, and a fresh approach to sales and marketing are all opportunities that the pharmaceutical sector may use. This condition of things is due to several variables. The pharmaceutical sector has certain distinct characteristics that influence product development.
“Fundamentally, however, the touchstone of FDA drug approval is a favorable benefit-risk assessment; without that favorable assessment, the drug should not have the status of being FDA-approved.” For additional information, see Makena Information on FDA.gov. Source link: [link]
In 2016, the Food and Drug Administration (FDA) approved Spinraza (nusinersen). While the FDA’sapproval of nusinersen may not seem extraordinary, it was. Nusinersen’s approval marked the first time nonclinical data supported conducting initial clinical trials involving children. Why This Guidance Now?
This feedback prompted the FDA to release a second paper in 2021 titled “ Artificial Intelligence/Machine Learning (AI.ML)-Based Software as a Medical Device (SaMD) Action Plan ,” which outlined the agency’s five-pillar approach to regulating AI/ML.
The treatment had been granted breakthrough therapy designation, but FDAapproval would ultimately rest on the sponsor’s ability to demonstrate clinically meaningful improvement. Key Takeaway RWD and RWE play an important role in rare diseases, where capturing relevant data to support drug development is challenging.
Researcher Considerations for IRB Review of Study Changes Modification or amendment information should include sufficient detail for IRB assessment. The more information researchers provide in the IRB submission, the easier it is for the IRB to consider implications for the research, researcher, and current and future research participants.
Molecular Weight: 631.700 FDAAPPROVED, To treat moderately to severely active ulcerative colitis in adults, 10/12/2023 Velsipity Etrasimod , sold under the brand name Velsipity , is a medication that is used for the treatment of ulcerative colitis (UC). [1] “FDAApproves New Drug for Ulcerative Colitis” Medscape.
The State’s claims violate abecedarian principles of due process by seeking to hold a company liable for conduct permitted under civil law and regulations. Food and Drug Administration (FDA)-approved and since launch, have reckoned for lower than one percent of total opioid conventions in Oklahoma as well as the United States.
BY AMANDA CONTI | JUL 24, 2024 9:57 PM CDT Background: Modernizing regulation of over-the-counter (OTC) products Nonprescription products, also called over-the-counter (OTC) products, must demonstrate the ability to be used safely and effectively without the supervision of a qualified healthcare professional.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.














































Let's personalize your content