This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
FDAApproves Sesquient (fosphenytoin sodium) for the Treatment of Status Epilepticus in Adult and Pediatric Patients. Food and Drug Administration (FDA) has approved Sesquient (fosphenytoin sodium for injection) for the treatment of status epilepticus in adult and pediatric patients. About Sedor Pharmaceuticals, LLC.
The Countering Emerging Threats - Rapid Acquisition and Investigation of Drugs for Repurposing (CET RAIDR) program within the JPM Medical is designed to rapidly tackle known, unknown, and emerging threats by utilizing late-stage or licensed therapeutics. Repurposing is one such method.
NYSE: PFE) and BioNTech SE (Nasdaq: BNTX) today announced they have submitted a supplemental Biologics License Application (sBLA) to the U.S. Food and Drug Administration (FDA) to expand the approval of COMIRNATY® (COVID-19 Vaccine, mRNA) to include individuals ages 12 through 15 years. Pfizer Inc.
The Pfizer-BioNTech COVID-19 Vaccine has not been approved or licensed by the U.S. Food and Drug Administration (FDA), but has been authorized for emergency use by FDA under an Emergency Use Authorization (EUA) to prevent Coronavirus Disease 2019 (COVID-19) for use in individuals 16 years of age and older.
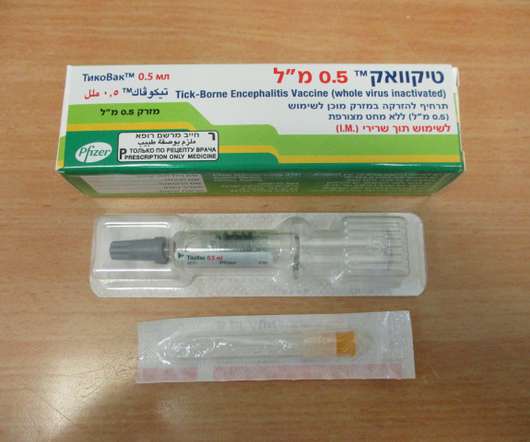
Food and Drug Administration (FDA) has approved TICOVAC (tick-borne encephalitis (TBE) vaccine) for active immunization to prevent TBE in individuals 1 year of age and older. 1 TICOVAC is the only FDA-approved vaccine to help protect U.S. Following today’s FDAapproval, the U.S.
FDAApproves Danyelza (naxitamab-gqgk) for the Treatment of Neuroblastoma. the “Company” or “Y-mAbs”) (Nasdaq: YMAB) a commercial-stage biopharmaceutical company focused on the development and commercialization of novel, antibody-based therapeutic products for the treatment of cancer, today announced that the U.S. NEW YORK, Nov.
Valentine — On November 22, 2022, FDAapproved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
Germany-based Cevec Pharmaceuticals GmbH signed a licensing agreement with Biogen for the use of its proprietary ELEVECTA Technology for the manufacturing of adeno-associated virus (AAV) vectors for gene therapy applications. The deal will provide Biogen the rights to use the technology across their portfolio of gene therapy products.
During the meeting, the FDA provided encouraging feedback regarding the Phase 3 study of omidubicel pertaining to the pre-specified primary and secondary endpoints. The FDA also recommended that Gamida Cell generate additional manufacturing-related data prior to requesting a pre-Biologics License Application (BLA) meeting.
Food and Drug Administration (FDA) to market Ephedrine Sulfate Injection in a ready-to-use 50mg/10 ml single use vial presentation. Under an exclusive licensing agreement with Endo International’s (NASDAQ: ENDP) subsidiary, Endo Ventures Limited, Par Pharmaceuticals’ Sterile Products division will launch and distribute the product.
FDAApproves Zokinvy (lonafarnib) for Hutchinson-Gilford Progeria Syndrome and Processing-Deficient Progeroid Laminopathies. Eiger licensed exclusive worldwide rights to lonafarnib from Merck, known as MSD outside of the United States and Canada. PALO ALTO, Calif., 20, 2020 /PRNewswire/ — Eiger BioPharmaceuticals, Inc.
Food and Drug Administration (FDA) approval of Darzalex Faspro ® (daratumumab and hyaluronidase-fihj), a subcutaneous formulation of daratumumab, in combination with bortezomib, cyclophosphamide and dexamethasone (D-VCd) for the treatment of adult patients with newly diagnosed light chain (AL) amyloidosis.[1]
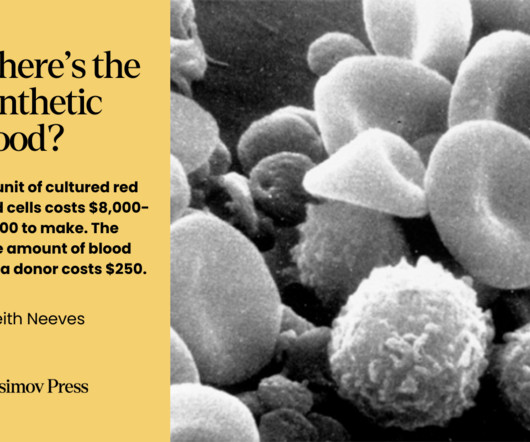
receives a blood transfusion or a blood product every two seconds. But currently, traditional blood transfusions face significant challenges, including a dwindling pool of donors and a limited shelf life for stored blood products. According to the American Red Cross, someone in the U.S.
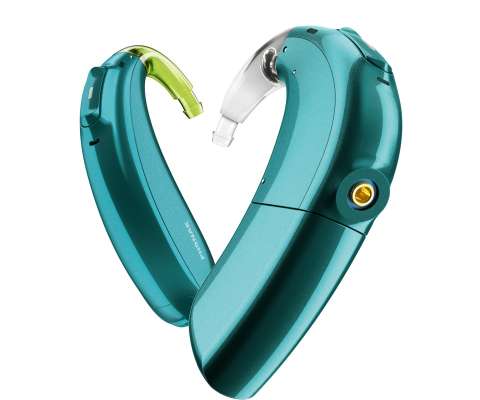
.–( BUSINESS WIRE )– Advanced Bionics (AB) , a global leader in cochlear implant technology, in collaboration with Phonak, a leading provider of life-changing hearing solutions, receives FDAapproval and announces it is bringing Marvel hearing technology to Advanced Bionics cochlear implant wearers. Life is on. Gifford, R.
NASDAQ: AUPH / TSX:AUP) (“Aurinia” or the “Company”) today announced it has entered into a collaboration and license agreement with Otsuka Pharmaceutical Co., The Company is currently seeking FDAapproval of voclosporin for the potential treatment of LN. VICTORIA, British Columbia & ROCKVILLE, Md.–(
CNS holds a worldwide exclusive license to the Berubicin chemical compound and has acquired all data and know-how from Reata Pharmaceuticals, Inc. The Company will initiate its trial during the first quarter of 2021 to investigate the efficacy of Berubicin in adults with GBM who have failed first-line therapy.
1, 2020 /PRNewswire/ — Sosei Group Corporation (“the Company”) (TSE: 4565) announces it has entered into a global collaboration and license agreement with Biohaven Pharmaceutical Holding Company Ltd. (“Biohaven”, NYSE: BHVN). .
TOKYO and CAMBRIDGE, England , Dec. Vlad Coric , M.D.,
Food and Drug Administration (FDA) has accepted for review the Prior Approval Supplement (PAS) to the Biologics License Application (BLA) for ABRILADA™ (adalimumab-afzb) as an interchangeable biosimilar to Humira® (adalimumab). The Biosimilar User Fee Act (BsUFA) goal date for an FDA decision is in Q4 2022. Pfizer Inc.
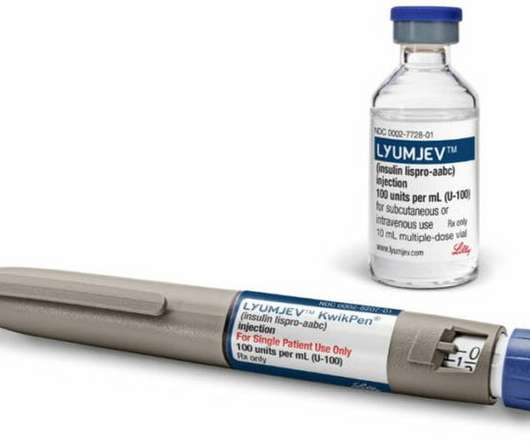
If your doctor decides to give your child any insulin products, he or she may give you special instructions. All Lyumjev and Humalog products contain insulin lispro. Your doctor may need to change or stop treatment with TZDs and your insulin lispro product.
WAKIX is the first and only treatment approved by the FDA for people with excessive daytime sleepiness or cataplexy associated with narcolepsy that is not scheduled as a controlled substance by the U.S. WAKIX received FDAapproval for the treatment of excessive daytime sleepiness in adult patients with narcolepsy in August 2019.
Food and Drug Administration (FDA) has approved PREVNAR 20 (Pneumococcal 20-valent Conjugate Vaccine) for the prevention of invasive disease and pneumonia caused by the 20 Streptococcus pneumoniae (pneumococcus) serotypes in the vaccine in adults ages 18 years and older. Following today’s FDAapproval, the U.S.
1] [2] It was developed by Vertex Pharmaceuticals , [5] and was approved for medical use in the United States in January 2025. [2] 2] [6] Suzetrigine is the first medication to be approved by the US Food and Drug Administration (FDA) in this new class of pain management medicines. [2] Food and Drug Administration (FDA).
FDA Actions. FDAApproval: Last week the FDAapproved Veklury (remdesivir) for the treatment of COVID-19 requiring hospitalization in adults and pediatric patients (12 years of age and older). News information is not all-inclusive and updates are published once a week on Tuesdays. . . Other Industry News.
Food and Drug Administration (FDA) has approved the company’s supplemental Biologics License Application for Xolair® (omalizumab) prefilled syringe for self-injection across all approved U.S. Roche’s Chief Medical Officer and Head of Global Product Development. with Xolair since its initial approval in 2003.
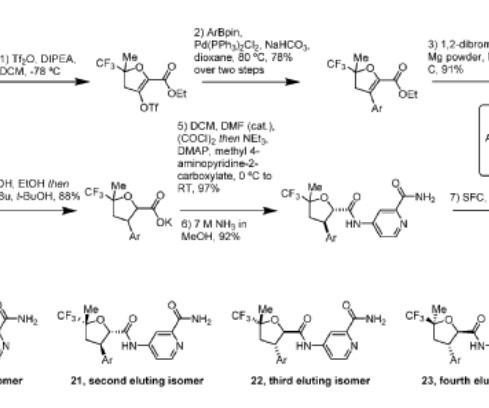
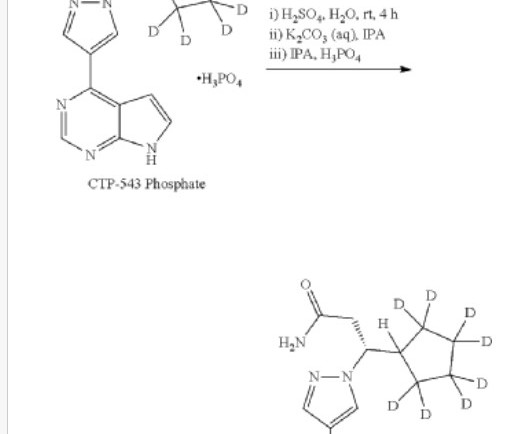
Deuruxolitinib C 17 H 18 N 6 , 314.422 Fdaapproved Leqselvi , 7/25/2024, To treat severe alopecia areata C-21543, CTP 543, CTP-543, CTP543 (3r)-3-(2,2,3,3,4,4,5,5-d8)cyclopentyl-3-(4-(7h-pyrrolo(2,3-d)pyrimidin-4-yl)-1h-pyrazol-1-yl)propanenitrile 1h-pyrazole-1-propanenitrile,beta.-(cyclopentyl-2,2,3,3,4,4,5,5-d8)-4-(7h-pyrrolo(2,3-d)pyrimidin-4-yl)-,
Rescheduling Analyses, 2016 DEA noted in 2016 that a drug had a currently accepted medical use for purposes of the CSA if it was the subject of an approved new drug application (“NDA”) under the Federal Food, Drug and Cosmetic Act. FDA, Basis for the Recommendation to Reschedule Marijuana Into Schedule III of the Controlled Substances Act, 3.
FDA advisory committees recommended just 50 percent of the 18 new therapies and indications they reviewed in 2020, the lowest rate since 2007, and the agency seems to be reserving the panels for more problematic applications, according to Prevision Policy, a Washington, D.C.-based based research firm.
1] Palopegteriparatide was approved for medical use in the European Union in November 2023, [2] and in the United States in August 2024. [1] 5] The FDA granted the application for palopegteriparatide orphan drug and priority review designations. [5] 4] [6] The applicant for this medicinal product is Ascendis Pharma Bone Diseases A/S.
3] In November 2023, capivasertib was approved in the United States for people with hormone receptor-positive, human epidermal growth factor receptor 2 -negative breast cancer when used in combination with fulvestrant. [3] The organic layer was dried over MgSO 4 , filtered and evaporated to afford crude product. 1H NMR (399.9
Lilly is offering donations of baricitinib to the Indian government through Direct Relief while simultaneously working with local Indian pharmaceutical companies to execute royalty-free voluntary licensing agreements to accelerate the manufacturing and distribution of the medicine in India during the pandemic. It is approved in the U.S.
Athenex and Almirall have a license deal with Almirall having the license to research, develop and commercialize the drug in the U.S. The FDAapproval of Klisyri is a significant milestone for Athenex,” said Johnson Lau, chairman and chief executive officer of Athenex. and Europe, including Russia. Most Read Today.
Amongst other things, FDA co-opted many of the same definitions for key terms for implementation of the BPCIA. FDA borrowed this definition from 21 C.F.R. mg/mL) for liquid parenteral drug products. mg/mL) for liquid parenteral drug products. mL) in addition to Original Concentration Humira.”
As a reminder, in Title I of the 2013 Drug Quality and Security Act (DQSA) (the Compounding Quality Act), Congress created the “outsourcing facility” FDA registration category, and set forth statutory parameters for their operation in new section 503B of the FDCA. See 21 U.S.C. 353b(a). Section II at 2. Draft Guidance III.B.2(e)
2] Vorasidenib was approved for medical use in the United States in August 2024. [2] 2] [3] It is the first approval by the US Food and Drug Administration (FDA) of a systemic therapy for people with grade 2 astrocytoma or oligodendroglioma with a susceptible isocitrate dehydrogenase-1 or isocitrate dehydrogenase-2 mutation. [2]
TERMS OF THE ARRANGEMENT Biohaven and Pfizer are entering into a collaboration and license agreement and affiliated sublicense agreement pursuant to which Pfizer will acquire rights to manipulate rimegepant and zavegepant outside of theU.S. In addition to the tiered double- number royalties owed to Biohaven on net deals outside of theU.S.,
Second FDAapproved indication for dostarlimab in 2021 GARNET study demonstrated objective response rate of 41.6% This indication received accelerated approval based on tumour response rate and durability of response. This approval was based on data from cohort A1, which included 71 patients with dMMR endometrial cancer. [vii]
If approved, Actemra/RoActemra would be the first U.S. FDAapproval is expected in the second half of this year. Chief Medical Officer and Head of Global Product Development, Roche. “The Chief Medical Officer and Head of Global Product Development, Roche. today announced that the U.S. A decision on U.S.
Phase 3 results evaluating the third dose are expected shortly and will be submitted to the FDA, the EMA and other regulatory authorities worldwide. Submissions to pursue regulatory approvals in those countries where emergency use authorizations or equivalent were initially granted are ongoing or planned. In the U.S.,
Y-mAbs Therapeutics has a target action date of November 30 for its Biologics License Application (BLA) for Danyelza (naxitamab) for patients with relapsed/refractory high-risk neuroblastoma. The drug was developed by researchers at Memorial Sloan Kettering Cancer Center and exclusively licensed to Y-mAbs.
156, as added by the 1984 Hatch-Waxman Amendments, for certain FDA-regulated products. The use of a surrogate endpoint can considerably shorten the time required prior to receiving FDAapproval. Karst — Earlier this week, we posted Part 1 of our three-part series on U.S.
Updated guidance on promotional labeling for biosimilars and interchangeables emphasizes a similar approach Today, the FDA issued a revised draft guidance on the development of promotional labeling for biosimilars, reference products, and—newly—interchangeable products.
Teva Branded Pharmaceutical Products R&D, Inc. NYSE and TASE: TEVA), have reached an agreement with Lupin to resolve the dispute over Lupin’s Abbreviated New Drug Application (“ANDA”) for a generic deutetrabenazine product. AUSTEDO is the first and only vesicular monoamine transporter 2 (VMAT2) inhibitor approved by the U.S.
As such, it doesn’t review things like vaccines, blood products or gene therapies – those products are instead reviewed by CBER. All products are received in one of two forms: A New Drug Application (or NDA, for pharmaceuticals) or a Biologics License Application (BLA, for biologics).
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.










































Let's personalize your content