This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
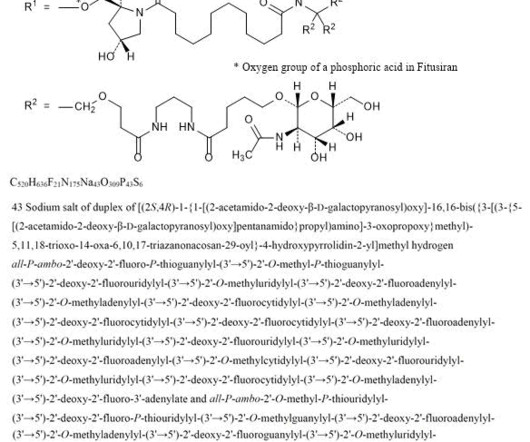
2] Fitusiran was approved for medical use in the United States in March 2025. [2] 2] The fixed dose of fitusiran is not approved because it led to excessive clotting in some participants. [2] 2] The US Food and Drug Administration (FDA) granted the application for fitusiran orphan drug and fast track designations. 26 March 2025.
The Countering Emerging Threats - Rapid Acquisition and Investigation of Drugs for Repurposing (CET RAIDR) program within the JPM Medical is designed to rapidly tackle known, unknown, and emerging threats by utilizing late-stage or licensed therapeutics. Repurposing is one such method.
The Pfizer-BioNTech COVID-19 Vaccine has not been approved or licensed by the U.S. Food and Drug Administration (FDA), but has been authorized for emergency use by FDA under an Emergency Use Authorization (EUA) to prevent Coronavirus Disease 2019 (COVID-19) for use in individuals 16 years of age and older.
FDAApproves Danyelza (naxitamab-gqgk) for the Treatment of Neuroblastoma. the “Company” or “Y-mAbs”) (Nasdaq: YMAB) a commercial-stage biopharmaceutical company focused on the development and commercialization of novel, antibody-based therapeutic products for the treatment of cancer, today announced that the U.S. NEW YORK, Nov.
Valentine — On November 22, 2022, FDAapproved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
During the meeting, the FDA provided encouraging feedback regarding the Phase 3 study of omidubicel pertaining to the pre-specified primary and secondary endpoints. The FDA also recommended that Gamida Cell generate additional manufacturing-related data prior to requesting a pre-Biologics License Application (BLA) meeting.
Food and Drug Administration (FDA) to market Ephedrine Sulfate Injection in a ready-to-use 50mg/10 ml single use vial presentation. Under an exclusive licensing agreement with Endo International’s (NASDAQ: ENDP) subsidiary, Endo Ventures Limited, Par Pharmaceuticals’ Sterile Products division will launch and distribute the product.
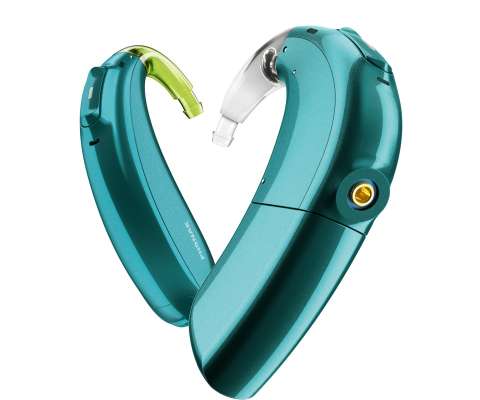
.–( BUSINESS WIRE )– Advanced Bionics (AB) , a global leader in cochlear implant technology, in collaboration with Phonak, a leading provider of life-changing hearing solutions, receives FDAapproval and announces it is bringing Marvel hearing technology to Advanced Bionics cochlear implant wearers. Life is on. Gifford, R.
CNS holds a worldwide exclusive license to the Berubicin chemical compound and has acquired all data and know-how from Reata Pharmaceuticals, Inc. The Company will initiate its trial during the first quarter of 2021 to investigate the efficacy of Berubicin in adults with GBM who have failed first-line therapy.
1, 2020 /PRNewswire/ — Sosei Group Corporation (“the Company”) (TSE: 4565) announces it has entered into a global collaboration and license agreement with Biohaven Pharmaceutical Holding Company Ltd. (“Biohaven”, NYSE: BHVN). .
TOKYO and CAMBRIDGE, England , Dec. Vlad Coric , M.D.,
1] [2] It was developed by Vertex Pharmaceuticals , [5] and was approved for medical use in the United States in January 2025. [2] 2] [6] Suzetrigine is the first medication to be approved by the US Food and Drug Administration (FDA) in this new class of pain management medicines. [2] Food and Drug Administration (FDA).
Food and Drug Administration (FDA) has approved the company’s supplemental Biologics License Application for Xolair® (omalizumab) prefilled syringe for self-injection across all approved U.S. Roche’s Chief Medical Officer and Head of Global Product Development. with Xolair since its initial approval in 2003.
FDA advisory committees recommended just 50 percent of the 18 new therapies and indications they reviewed in 2020, the lowest rate since 2007, and the agency seems to be reserving the panels for more problematic applications, according to Prevision Policy, a Washington, D.C.-based based research firm.
1] Palopegteriparatide was approved for medical use in the European Union in November 2023, [2] and in the United States in August 2024. [1] 5] The FDA granted the application for palopegteriparatide orphan drug and priority review designations. [5] 4] [6] The applicant for this medicinal product is Ascendis Pharma Bone Diseases A/S.
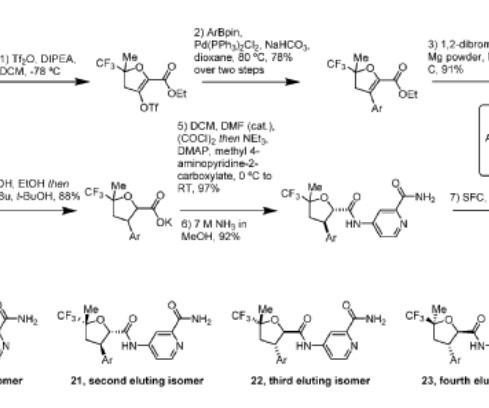
3] In November 2023, capivasertib was approved in the United States for people with hormone receptor-positive, human epidermal growth factor receptor 2 -negative breast cancer when used in combination with fulvestrant. [3] The organic layer was dried over MgSO 4 , filtered and evaporated to afford crude product. 1H NMR (399.9
Pfizer plans to file for full FDAapproval of Covid vaccine at the end of this month ( CNBC ).
The FDA is set to authorize the Pfizer-BioNTech vaccine for those 12-15 years old by early next week.
FDAapproves expanded use for Chiesi’s sickle cell drug Ferriprox ( PMLive ).
That’s why, according to Salant, drug companies “[blur] the distinction between dangerous counterfeit drugs and safe drugs sold by pharmacies licensed in other high-income countries.” Third, even foreign drug prices are much higher than the cost of production. The paper makes allowances only for imports from high-income countries.
Amongst other things, FDA co-opted many of the same definitions for key terms for implementation of the BPCIA. FDA borrowed this definition from 21 C.F.R. mg/mL) for liquid parenteral drug products. mg/mL) for liquid parenteral drug products. mL) in addition to Original Concentration Humira.”
As a reminder, in Title I of the 2013 Drug Quality and Security Act (DQSA) (the Compounding Quality Act), Congress created the “outsourcing facility” FDA registration category, and set forth statutory parameters for their operation in new section 503B of the FDCA. See 21 U.S.C. 353b(a). Section II at 2. Draft Guidance III.B.2(e)
2] Vorasidenib was approved for medical use in the United States in August 2024. [2] 2] [3] It is the first approval by the US Food and Drug Administration (FDA) of a systemic therapy for people with grade 2 astrocytoma or oligodendroglioma with a susceptible isocitrate dehydrogenase-1 or isocitrate dehydrogenase-2 mutation. [2]
If approved, Actemra/RoActemra would be the first U.S. FDAapproval is expected in the second half of this year. Chief Medical Officer and Head of Global Product Development, Roche. “The Chief Medical Officer and Head of Global Product Development, Roche. today announced that the U.S. A decision on U.S.
Phase 3 results evaluating the third dose are expected shortly and will be submitted to the FDA, the EMA and other regulatory authorities worldwide. Submissions to pursue regulatory approvals in those countries where emergency use authorizations or equivalent were initially granted are ongoing or planned. In the U.S.,
Y-mAbs Therapeutics has a target action date of November 30 for its Biologics License Application (BLA) for Danyelza (naxitamab) for patients with relapsed/refractory high-risk neuroblastoma. The drug was developed by researchers at Memorial Sloan Kettering Cancer Center and exclusively licensed to Y-mAbs.
156, as added by the 1984 Hatch-Waxman Amendments, for certain FDA-regulated products. The use of a surrogate endpoint can considerably shorten the time required prior to receiving FDAapproval. Karst — Earlier this week, we posted Part 1 of our three-part series on U.S.
Updated guidance on promotional labeling for biosimilars and interchangeables emphasizes a similar approach Today, the FDA issued a revised draft guidance on the development of promotional labeling for biosimilars, reference products, and—newly—interchangeable products.
Teva Branded Pharmaceutical Products R&D, Inc. NYSE and TASE: TEVA), have reached an agreement with Lupin to resolve the dispute over Lupin’s Abbreviated New Drug Application (“ANDA”) for a generic deutetrabenazine product. AUSTEDO is the first and only vesicular monoamine transporter 2 (VMAT2) inhibitor approved by the U.S.
, including inquires from additional potential international distributors as well as discussions with several of its large GenUltimate distributors who have expressed interest in having, after FDA authorization, our GenViro! added to their Amazon professional use product offerings,” said CEO Keith Berman.
NYSE American: PLX) (TASE: PLX), a biopharmaceutical company focused on the development, production and commercialization of recombinant therapeutic proteins produced by its proprietary ProCellEx ® plant cell-based protein expression system, and Chiesi Global Rare Diseases, a business unit of Chiesi Farmaceutici S.p.A.,
The booster schedule is based on the labeling information of the vaccine used for the primary series COMIRNATY® (COVID-19 Vaccine, mRNA) is an FDA-approved COVID-19 vaccine made by Pfizer for BioNTech. Pfizer Inc.
The FDA has also granted precedence review to the company’s sBLA for Kymriah in adult cases with r/ r FL. The decision follows a positive opinion from the Committee for Orphan Medicinal Products ( Presentation) of the EMA. About people of further than 140 ethnicities work at Novartis around the world.
To date, Brilacidin has received Fast Track designation for three different clinical indications: COVID-19, Oral Mucositis, and Acute Bacterial Skin and Skin Structure Infection (qualifying for Fast Track under Qualified Infectious Disease Product designation). Brilacidin for UP/UPS was licensed to Alfasigma S.p.A.
Now poised to advance a robust therapeutics pipeline to clinical development, Nuance will use the funds for ongoing R&D of existing products and business development of potential new assets. Pear’s reSET, reSET-O and Somryst are the first PDTs to receive FDAapproval for treating disease. Sigilon Therapeutics .
Founded and led for over 30 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to eight FDA-approved treatments and numerous product candidates in development, all of which were homegrown in our laboratories.
It increases the optionality to the Company’s value proposition, leverages our endocrine expertise and adds a late-stage pivotal trial-ready orphan disease product to the existing Phase 3 programs for abaloparatide and elacestrant,” said Radius Chief Executive Officer, Kelly Martin. RAD011 has the potential for broad clinical applicability.
The application will be reviewed by the EMA’s Committee for Medicinal Products for Human Use (CHMP) under the centralized licensing procedure for all 27 Member States of the European Union, as well as Norway, Iceland and Liechtenstein. Additional data from the CAPELLA study will be presented at a future scientific conference.
Food and Drug Administration (FDA) for the treatment of mild to moderate COVID-19 in patients 12 years of age and older and weighing at least 40 kg, who have received positive results of direct SARS-CoV-2 viral testing and are at high risk for progressing to severe COVID-19 and/or hospitalization.
Pfizer and BioNTech expect to file a Biologics License Application for possible full regulatory approval in 2021.
Food and Drug Administration (FDA) has authorized the emergency use of the mRNA vaccine, BNT162b2, against COVID-19 in individuals 16 years of age or older.
Food and Drug Administration (FDA) has accepted for review the supplemental Biologics License Application (sBLA) for Dupixent ® (dupilumab) as an add-on treatment for children aged 6 to 11 years with uncontrolled moderate-to-severe asthma.
Nasdaq: MRNA), a biotechnology company pioneering messenger RNA (mRNA) therapeutics and vaccines, today announced that Swissmedic, the Swiss Agency for Therapeutic Products, has authorized the COVID-19 Moderna Vaccine in Switzerland. MRNA-1273 (SARS-CoV-2 vaccine) FDAApproval History. .–(BUSINESS WIRE)–Jan.
NYSE American:PLX) (TASE:PLX), a biopharmaceutical company focused on the development, production and commercialization of recombinant therapeutic proteins produced by its proprietary ProCellEx ® plant cell-based protein expression system, and Chiesi Global Rare Diseases , a business unit of Chiesi Farmaceutici S.p.A.,
resolving patent litigation brought in response to Teva’s Abbreviated New Drug Application, seeking approval to market a generic version of Xtampza ER prior to the expiration of Collegium’s applicable patents. FDAapproval, and customary exceptions). Xtampza ER net product revenues were $32.1
The study, which analyzed vast troves of genomic and clinical data collected over many years from more than 50,000 people with and without diabetes, indicates that anti-diabetes therapies that lower glucose by targeting the product of a specific gene, called GLP1R , are unlikely to boost the risk of cardiovascular disease.
–( BUSINESS WIRE )– Bristol Myers Squibb (NYSE: BMY) today announced that the Biologics License Application (BLA) for lisocabtagene maraleucel (liso-cel) for the treatment of adults with relapsed or refractory (R/R) large B-cell lymphoma after at least two prior therapies remains under review by the U.S. 1, 2021 11:59 UTC.
The EMA and the Committee for Medicinal Products for Human Use reviewers, working over the holidays, provided a thorough review and comprehensive guidance as we worked together to achieve this authorization. MRNA-1273 (SARS-CoV-2 vaccine) FDAApproval History. supply chain are expected to begin next week. Posted: January 2021.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.
















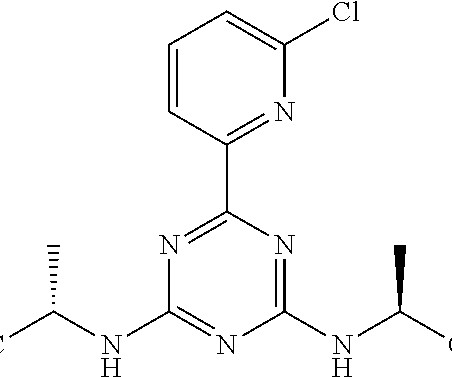





























Let's personalize your content