This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Further, combined with the early successes in improving cardiac performance, now when combined with MRI compatibility, CCM adds to the armamentarium of the cardiologist and electrophysiologist in further optimizing FDA-approved medical therapies in 2021.”
But FDA-approved forms of the most commonly used vehicle for packaging and delivering these therapies to target cells, adeno-associated viruses (AAVs), aren’t able to efficiently cross the blood-brain barrier at high levels and deliver therapeutic cargo.
Dive into this week’s update for more details on the actions taken by the FDA in the ongoing response to the Covid-19 pandemic. FDAapproves first treatment for Covid-19. On October 22, the FDAapproved the antiviral drug Veklury for use in adult and pediatric patients for the treatment of Covid-19 requiring hospitalization.
Valentine — On November 22, 2022, FDAapproved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
By Allessandra DiCorato July 19, 2023 Credit: Kevin Middleton, Broad Communications A three-dimensional model of adeno-associated viruses (AAVs), which scientists have engineered to package and deliver gene therapies to cells in the body.
A significant effort has been made to correctly map the drug form of the EMA data by manually inspecting different EMA sources of information, such as the Product Information (Annex I: Summary of Product Characteristics and Annex III: Labelling and Package Leaflet) and/or Assessment Report, where available. University of Dundee: T.
Effect on FDAapprovals— In cases where a biological product or drug needs to change aspects of its manufacturing processes to avoid using a covered equipment or service, will it need to file supplements with FDA for the CMC update?
When returned directly to participants, aggregate results should be packaged and communicated in user-friendly formats, such as in newsletters or web pages constructed with readability and health literacy principles in mind. Conversely, a CGM not yet FDA-approved assessed in the study for reliability, would yield an investigational result.
These initiatives could support insurance coverage of previously off-label uses, prevent costly new drugs from inappropriately receiving preferential regulatory treatment that is intended for drugs with no FDA-approved alternatives, and better inform clinicians by providing evidence-based information about how drugs should be used,” they noted. .
Limited Evidence for Nalfmefene "In 2021, due to the widespread availability of high-potency synthetic opioids like fentanyl, the US FDAapproved two high-dose naloxone products, an 8 mg IN spray (Kloxxado) and a 5 mg IM injectable (Zimhi). In 2023, the FDAapproved a 2.7
Pfizer is a leader in this vital healthcare segment with more than 14 years of global in-market experience, the first FDAapproved biosimilar for the treatment of certain autoimmune conditions and nine approved biosimilars in the U.S.
End-to-End Single-Site Solution from Drug Substances to Fill-Finish & Packaging. In addition to drug substance manufacture, the facility will also provide commercial scale, automated fill-finish and assembly, packaging and labeling services. Packaging line?.
TOKYO , Jan. 8 x 20,000L bioreactors for mammalian cells.
As part of the comprehensive submission package to the EMA, Sandoz conducted a Phase I pharmacokinetics (PK) bridging study comparing Hyrimoz 50 mg/mL 2 and Hyrimoz HCF. The adalimumab reference medicine (Humira ®* ) was first approved with an adalimumab concentration of 50 mg/mL.
However, the success of a 505(b)(2) application hinges on a tailored development strategy that carefully considers the specific characteristics of the newly proposed drug product, and the nature of the changes made in comparison to a prior US FDA-approved listed drug (LD) or the drug reported in literature.
To this point, Moderna has only submitted two months of follow-up safety data, and the FDA typically requires six months for a full approval. intends to ship just shy of six million doses of Moderna’s vaccine once the FDAapproves EUA. Initial doses are expected to be limited as manufacturing ramps up.
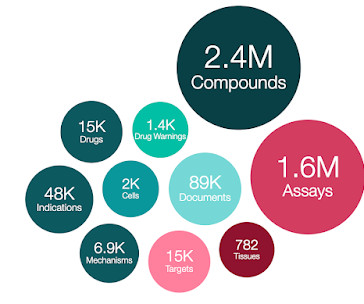
ChEMBL database version ChEMBL 33 release notes _ # This version of the database, prepared on 31/05/2023 contains: 2,399,743 compounds (of which 2,372,674 have mol files) 3,051,613 compound records (non-unique compounds) 20,334,684 activities 1,610,596 assays 15,398 targets 88,630 documents BioAssay Data Sources: Number Assays: Number Compound Records: (..)
Even for repurposed drugs being developed under the 505(b)(2) New Drug Application (NDA) pathway, it is critical to review the existing nonclinical and clinical data on the drug to determine what nonclinical studies may be beneficial to conduct prior to the PIND meeting and include this information in the package.
The product is made here, in the USA, in our FDAapproved and GMP certified facility, under sterile, strict and precise standards. After you fill in your information and confirm your package will be shipped for free right to your doorstep as soon as possible. Dentitox Pro is non-GMO and safe.
The treatment had been granted breakthrough therapy designation, but FDAapproval would ultimately rest on the sponsor’s ability to demonstrate clinically meaningful improvement. The FDA also granted a second meeting for review. The agency encouraged further review of the data for potential reference as a historical control.
A: This is a complex topic and is best outlined in the FDA’s guidance document. Q: What is the typical time period between the submission of the briefing package and the pre-IND meeting? A: The FDA’s guidance document indicates the briefing package is submitted four weeks before the meeting. Could you please confirm?
Instead of the black, printed stripes of the Universal Product Codes (UPCs) that we see on everything from package deliveries to clothing tags, they used short, unique snippets of DNA to label cells. A little more than a decade ago, researchers began adapting a familiar commercial concept to genomics: the barcode.
Food and Drug Administration – labeled an “Official Action Indicated” classification – is generally devastating for the facility, not least because it can stall FDAapproval of applications to market drugs manufactured at the facility. Farquhar — A drug manufacturer’s bad post-inspection grade from the U.S.
As part of the comprehensive submission package to the European Marketing Authorization, Sandoz conducted a Phase I pharmacokinetics (PK) bridging study comparing its approved adalimumab 50 mg/mL 2 with the 100 mg/mL (HCF). The adalimumab reference medicine (Humira ® *) was first approved with an adalimumab concentration of 50 mg/mL.
” AstraZeneca has already submitted a substantial data package to support a conditional marketing authorisation for its COVID-19 vaccine to the European Medicines Agency (EMA), as part of an ongoing rolling review process and will continue to work closely with the EMA to seek approval in the coming weeks. Source: AstraZeneca.
Once samples are validated, this will conclude the IND package” said Kyle Klepner, Senior Scientist, Study Director, Safety Assessment, at Altasciences. Virpax is initially seeking FDAapproval for two prescription drug candidates that employ two different patented drug delivery platforms.
Pear’s reSET, reSET-O and Somryst are the first PDTs to receive FDAapproval for treating disease. Already having been in clinical studies at Roche, all four candidates have strong clinical and preclinical safety packages. Series D funds will be used to accelerate reimbursement coverage for its three commercial products. .
The difference at issue here is the fact that Vanda’s Hetlioz product includes braille writing on its packaging, with some accompanying language (“Do not cover Braille”), and the generic products’ labeling does not. Vanda’s Hetlioz was, in fact, the first FDA-approved drug product to include braille labeling. . § 314.94(a)(8)(iv),
1 In 2017, the US Food and Drug Administration (FDA) approved the first AAV-based gene replacement therapy (Luxturna), for Leber congenital amaurosis type 2. 2 Since then, the FDA has approved four more AAV-based gene therapies—Zolgensma, Hemgenix, Elevidys and Rocktavian—for treating various diseases.
Prior to CARES Act reforms, FDA relied on a monograph process through which firms could bring OTC drugs to market without FDAapproval so long as they adhered to pre-set terms under the monograph. On the other hand, data, information and comments to a proposed or final order should use the OTC Monographs@FDA portal.
While the definition of NME has changed over the years, it can sometimes include a combination product consisting of at least one drug that has previously been approved. Data on these novel approvals is published throughout the year by both CDER and CBER. FDAapproved 13 NMEs through the AA pathway in FY 2023, making up 25.5%
Every capsule is made here, in the USA, in our FDAapproved and GMP certified facility, under sterile, strict and precise standards. If you order the 3 bottles or 6 bottles package (which we highly recommend as we estimate that we will run out of stocks soon as this has happened before) you’ll also take advantage of a huge discount.
Within the realm of FDA-required labeling, there are currently a few different types of information a sponsor might develop specifically for patient use: medication guides, instructions for use (IFU), consumer medical information (CMI) and patient package inserts (PPI). After all, companies already have FDA-approved labels.
Such a situation is commonplace in the clinical trial realm, in which investigational drug products which are not already FDAapproved are administered to patients. Under the Pandemic and All-Hazards Preparedness Reauthorization Act of 2013 (PAHPRA) , the FDA also has some authority to extend MCM expiration dates.
. | NOV 8, 2023 10:02 PM CST Regulatory background The Federal Food, Drug, and Cosmetics Act (FD&C Act) is administered by the Food and Drug Administration (FDA) and regulates cosmetics. MoCRA also requires all marketed cosmetic products to be listed with the FDA by December 29, 2023.
After all my requirements were 100% met, we finally had the final product: Every capsule is made here, in the USA, in our FDAapproved and GMP certified facility, under sterile, strict and precise standards.Met Slim Pro capsules are non-GMO and safe. Safe: processed under strict sterile standards with regularly disinfected equipment.
Casirivimab and imdevimab injection is an investigational combination therapy and has been authorized by FDA for the emergency use described above. Casirivimab and imdevimab injection is not FDAapproved for any use. REGN-COV2 (casirivimab and imdevimab) FDAApproval History. Source: Regeneron. Source link.
See “Worldwide Pro Forma Revenue” in Quarterly Package of Financial Information for this quarter, which is available on bms.com/investors/financial-reporting/quarterly-results, for information on the revenue of the company and Celgene on a stand-alone basis for the prior-year period. Oncology and Hematology. Regulatory. Regulatory.
The term “drug product information” is now defined to include NDC, drug name, units per package size (UPPS), drug category (S, I, or N), unit type (e.g.,
Every capsule is manufactured in the USA, in our FDAapproved and GMP certified facility, under sterile, strict and precise standards. If you order the 3 bottles or 6 bottles package (which we highly recommend as we estimate that we will run out of stocks soon as this has happened before) you’ll also take advantage of a huge discount.
In the case of semaglutide, those cells are Saccharomyces cerevisiae— also known as Baker’s yeast — engineered to secrete a peptide precursor that is later purified, chemically modified, packaged into an injectable or tablet form, and then shipped around the world. Continuing this method may not scale.
Founded and led for over 30 years by physician-scientists, our unique ability to repeatedly and consistently translate science into medicine has led to eight FDA-approved treatments and numerous product candidates in development, all of which were homegrown in our laboratories.
Authorized Emergency Use Casirivimab and imdevimab injection is an investigational combination therapy and has been authorized by FDA for the emergency use described above. Casirivimab and imdevimab injection is not FDAapproved for any use. 360bbb-3(b) (1), unless the authorization is terminated or revoked sooner.
Read Inhalable nanoparticles, packaged with mRNA or CRISPR systems, efficiently edit lung cells. Large-scale purification of functional AAV particles packaging the full genome using short-term ultracentrifugation with a zonal rotor. Nature Biotechnology. Gene Therapy. Press release. Press release.
Explain the FDAApproval Process Many patients are unaware of the rigorous approval process generic drugs must undergo. Educate them about the FDA’s role in ensuring the safety and efficacy of generic medications. These anecdotes can help alleviate fears and build confidence in generic medications.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.













































Let's personalize your content