This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
In general, existence of sp 3 -O slightly outperforms sp 2 -O, which is associated with balancing various factors such as flexibility, solubility, stability, and pharmacokinetics, in addition to activity and selectivity. In approved drugs, majority of oxygen atoms are present within 4 from the COM of the molecule.
Lenacapavir (LEN), a long-acting injectable, is the first approved human immunodeficiency virus type 1 capsid inhibitor and one of a few FDA-approved drugs that exhibit atropisomerism.
Together, the tools estimate how a drug may impact diverse outcomes of interest to drug developers: general cellular health, pharmacokinetics, and heart and liver function. Seal used these FDA-curated lists as training data for two toxicity-predicting machine learning models: one for cardiotoxicity and one for liver injury.
Secondary VAS and pharmacokinetic (PK) endpoints and adverse events were assessed. Drug Liking and all other VAS outcomes were greatest for nabilone 3mg and 6mg, which is a currently FDA-approved medication. Three doses of lenabasum (20, 60, and 120mg) were compared to placebo, and nabilone (3 and 6mg).
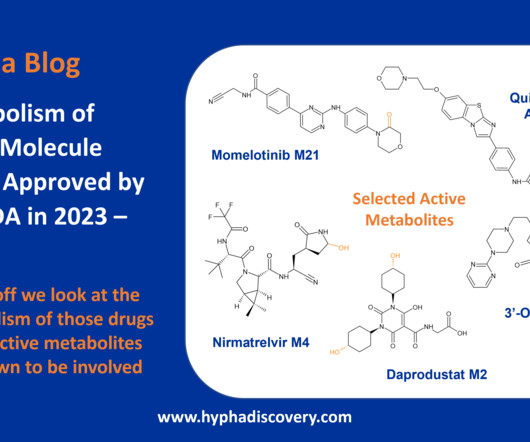
Evolution of Ritlecitinib Population Pharmacokinetic Models During Clinical Drug Development. Clin Pharmacokinet. link] The post Metabolism of 2023 FDAApproved Small Molecules – PART 2 appeared first on Hypha Discovery. link] [10] Wojciechowski J, S Purohit V, Huh Y, Banfield C, Nicholas T. 2023; 62(12):1765-1779.
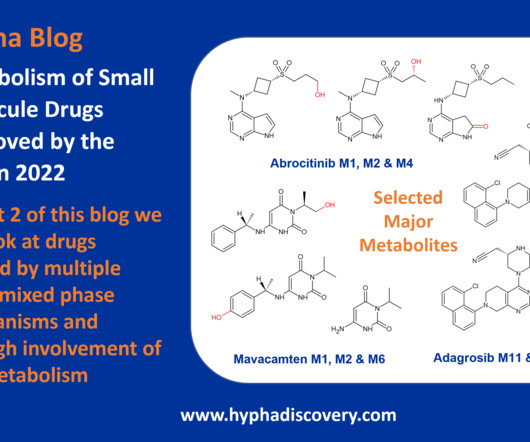
Metabolism of 2022 FDAapproved small molecule drugs part 2 Mixing it Up By Julia Shanu-Wilson In Part 1 of this topic we looked at metabolism of the small molecule drugs approved by the FDA in 2022 that were mediated by CYP3A4. We hope it was a useful two-parter! We’re looking forward to the next crop! Br J Pharmacol.
Metabolism of 2023 FDAApproved Small Molecules – PART 1 By Julia Shanu-Wilson 2023 was a fruitful year for drug approvals by the FDA, with a crop of 34 small molecules out of a total of 55 new drugs [1]. link] [10] FDA prescribing information for gepirone. Pharmacokinetics and Metabolism of Nirmatrelvir.
New Indication for Amgen’s Fifth FDA-approved Biosimilar. Now Approved to Treat All Available Rituxan ® Indications. Overall, 311 patients were randomized and treated with RIABNI, rituximab RP approved in the EU (rituximab-EU) or rituximab RP approved in the US (rituximab-US).
Food and Drug Administration (FDA) approved DALVANCE® (dalbavancin) for the treatment of acute bacterial skin and skin structure infections (ABSSSI) in pediatric patients from birth. “This pediatric approval for DALVANCE as a single-dose provides a meaningful contribution to the treatment of children and infants with ABSSSI.”
Submission supported by comprehensive analytical and clinical data from new Phase I bridging pharmacokinetics study Adalimumab’s high-concentration 100 mg/mL formulation aims to provide an enhanced yet familiar experience for patients Submission builds on Sandoz’ well established biosimilar immunology portfolio in Europe.
Even for repurposed drugs being developed under the 505(b)(2) New Drug Application (NDA) pathway, it is critical to review the existing nonclinical and clinical data on the drug to determine what nonclinical studies may be beneficial to conduct prior to the PIND meeting and include this information in the package.
There are many reasons that promising drug candidates are discontinued, including poor pharmacokinetics, lack of clinical efficacy, and toxicity. Furthermore, nearly one third of drugs get withdrawn from the market post approval due to safety concerns. link] New safety concerns identified for 1 in 3 FDA-approved drugs [Internet].
“Population pharmacokinetic/pharmacodynamic modelling of eplontersen, an antisense oligonucleotide in development for transthyretin amyloidosis” British Journal of Clinical Pharmacology. S2CID 250989659. ^ “Eplontersen: FDA-Approved Drugs” U.S. Food and Drug Administration (FDA). (December 2022).
As part of the comprehensive submission package to the European Marketing Authorization, Sandoz conducted a Phase I pharmacokinetics (PK) bridging study comparing its approved adalimumab 50 mg/mL 2 with the 100 mg/mL (HCF). The adalimumab reference medicine (Humira ® *) was first approved with an adalimumab concentration of 50 mg/mL.
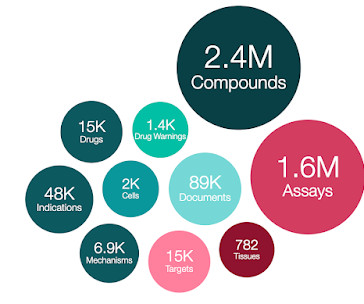
ChEMBL database version ChEMBL 33 release notes _ # This version of the database, prepared on 31/05/2023 contains: 2,399,743 compounds (of which 2,372,674 have mol files) 3,051,613 compound records (non-unique compounds) 20,334,684 activities 1,610,596 assays 15,398 targets 88,630 documents BioAssay Data Sources: Number Assays: Number Compound Records: (..)
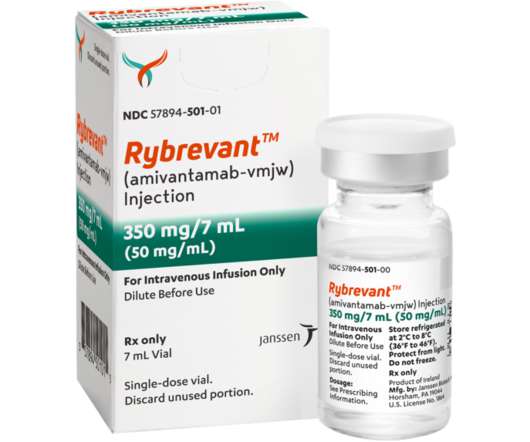
Food and Drug Administration (FDA) approval for patients with locally advanced or metastatic NSCLC with epidermal growth factor receptor (EGFR) exon 20 insertion mutations, based on data showing an ORR of 40 percent (95 percent CI, 29 – 51) and median duration of response of 11.1 months (95 percent CI, 6.9 – NE). [7].
of new cancer drugs tested in Phase I were likely to receive Food and Drug Administration (FDA) approval. Participants may balk at certain protocol requirements, such as extended visits for pharmacokinetic (PK) measurements. And if they do participate, they may be more likely to drop out prior to reaching a study endpoint.
Molecular Weight: 631.700 FDAAPPROVED, To treat moderately to severely active ulcerative colitis in adults, 10/12/2023 Velsipity Etrasimod , sold under the brand name Velsipity , is a medication that is used for the treatment of ulcerative colitis (UC). [1] “FDAApproves New Drug for Ulcerative Colitis” Medscape.
The FDAapproved the first gene therapy in 2017 for certain pediatric and young adult patients with a form of acute lymphoblastic leukemia (ALL), and as of December 2023, there are over 30 approved cell and gene therapies on the market in the U.S.A. Asia, and Europe.
In spite of current success and possibility to be a successful cell technology model, the integration of organ-on-chips into drug development process needs more optimisation to be validated for FDAapproval.
Because of its selectivity for oxycodone, the vaccine will not interfere with FDA-approved medications, including methadone, buprenorphine, naltrexone and naloxone, potentially offering a long-lasting, safe and cost-effective alternative that is complementary to standard medical intervention for opioid use disorders.
However, the success of a 505(b)(2) application hinges on a tailored development strategy that carefully considers the specific characteristics of the newly proposed drug product, and the nature of the changes made in comparison to a prior US FDA-approved listed drug (LD) or the drug reported in literature. Human factors.
This approach capitalizes on prior investments in R&D, mitigates risk by leveraging established safety and pharmacokinetic profiles, and accelerates the delivery of treatments to patients. The National Institutes of Health (NIH) Clinical Collection , a library of FDA-approved drugs, is widely used for HTS in drug repurposing initiatives.
There are currently no FDA-approved anticoagulation therapies for pediatric patients with congenital heart disease who have undergone the Fontan procedure. From November 2016 to June 2019, a total of 112 participants were enrolled across 36 sites in 10 countries. UNIVERSE was conducted in two parts.
About the FENopta study The FENopta study is a global Phase II, randomised, double-blind, placebo-controlled 12-week study to investigate the efficacy, safety and pharmacokinetics of fenebrutinib in 109 adults aged 18-55 years with RMS. Until the FDAapproval of OCREVUS, there had been no FDA-approved treatments for PPMS.
“We are pleased to continue pursuing additional neuroscience opportunities with BXCL501, targeting agitation associated with delirium, a fifth potential indication for this candidate and a condition for which there is no FDA-approved treatment,” commented Vimal Mehta, Chief Executive Officer of BTI.
Protein sub-units are covalently bound via chemical cross-linking using short PEG moieties, resulting in a molecule with unique pharmacokinetic parameters. Protalix was the first company to gain FDAapproval of a protein produced through plant cell-based in suspension expression system. Galactosidase-A enzyme.
Protein sub-units are covalently bound via chemical cross-linking using short PEG moieties, resulting in a molecule with unique pharmacokinetic parameters. Protalix was the first company to gain FDAapproval of a protein produced through plant cell-based in suspension expression system. Galactosidase-A enzyme.
Part B of the Phase 3 trial was informed by Part A , an open-label, single-ascending-dose, sequential cohort Phase 2 trial designed to assess the pharmacokinetics and safety of Dupixent in children aged 6 months to 5 years with uncontrolled severe atopic dermatitis.
Until recently, the FDA relied on a monograph process through which firms could bring OTC drugs to market without FDAapproval so long as it adhered to pre-set terms under the monograph. The committee also made recommendations regarding pharmacokinetic and safety assessments.
The Phase I/IIa clinical trial is a randomized, double-blind, placebo controlled, single and multiple dose escalation study to assess the safety, tolerability, pharmacokinetics and pharmacodynamics of NMD670 in 79 male and female healthy subjects and patients with myasthenia gravis. The secondary outcome involves pharmacokinetic endpoints.
Food and Drug Administration (FDA) approved EVRYSDI for the treatment of SMA in adults and children 2 months of age and older. EVRYSDI was granted PRIME designation by the European Medicines Agency (EMA) in 2018 and Orphan Drug Designation by FDA and EMA in 2017 and 2019, respectively.
Evaluation of the Pharmacokinetic Interaction and Safety of Coadministered Atogepant and Topiramate. About BOTOX ® BOTOX ® was first approved by the FDA in 1989 for two rare eye muscle disorders – blepharospasm and strabismus in adults. Abstract Lecture. September 12, 2021. 2:45-2:55 a.m. ePoster (on-demand only). Ubrogepant.
FDA-approved oral prescription medicine, 120 mg or 160 mg dependent on weight (<50 kg or ?50 i Continued approval may be contingent upon verification and description of clinical benefit in confirmatory trials. Retevmo may affect both tumor cells and healthy cells, which can result in side effects. Retevmo is an U.S.
Additionally, Regeneron bispecifics are manufactured using similar approaches used for human monoclonal antibody medicines, yielding similar properties and pharmacokinetics. These allow for the creation of bispecific antibodies that closely resemble natural human antibodies with no linkers or artificial sequences.
Hepatic Impairment: EDURANT ® should be used with caution in patients with severe hepatic impairment (Child-Pugh Class C) as pharmacokinetics of EDURANT ® have not been evaluated in these patients.
.
Please see full Prescribing Information for more details. Use in Specific Populations.
It also displayed favourable pharmacokinetics (PK) and is well tolerated in non-human primates (NHP) at exposure levels above those projected to be efficacious. ZW191 also displayed favourable pharmacokinetics (PK) and is well tolerated in non-human primates (NHP) at exposure levels above those projected to be efficacious.
It also provided supporting pharmacokinetic data demonstrating the opioid antagonist’s safety and efficacy. . In the case of Kloxxado, the applicant used the agency’s safety and effectiveness findings for Narcan (injectable naloxone hydrochloride) to support its application. New indications.
The FDAapproval of INVEGA HAFYERA™ is based on the results of a 12-month, randomized, double-blind, non-inferiority Phase 3 global study that enrolled 702 adults (ages 18-70) living with schizophrenia from 20 countries. 1 Study evaluations included efficacy, safety, pharmacokinetics, and pharmacodynamics.
For legacy AUC and Cmax records (< ChEMBL 34), pharmacokinetic parameters have been extracted from the assay descriptions using regular expression matching (RegEx). When possible, AUC record units were converted to ng.hr.mL-1 1 and Cmax record units were converted to nM by creating new conversion rules.
Nasdaq GILD) moment blazoned new data from an interim analysis of its ongoing, Phase2/3 single arm, open- marker study to estimate the safety, tolerability and pharmacokinetics of Veklury ® (remdesivir) in pediatric cases rehabilitated with COVID-19 with periods ranging from 28 days to lower than 18 times. Gilead Lores,Inc.
The study met its primary goal by demonstrating pharmacokinetic equivalence in patients who switched multiple times between treatment with the two medicines. The FDAapproval was based on the review of a comprehensive data package which demonstrated biosimilarity of ABRILADA to the reference product.
The Phase 1/2 trial aims to evaluate the safety, pharmacokinetics and anti-tumor activity of oral mobocertinib in patients with non-small cell lung cancer (NSCLC). Mobocertinib is the first oral therapy specifically designed to selectively target EGFR Exon20 insertion mutations. About the Phase 1/2 Trial.
TREMFYA q4w is not currently FDA-approved. Efficacy, safety, pharmacokinetic, immunogenicity and biomarker evaluations were performed in the study on a defined schedule. He has not been compensated for any media work. Clinically meaningful defined as ?3 About DISCOVER-2 (NCT03158285) 14. About Psoriatic Arthritis.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.





































Let's personalize your content