This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Learn more about FDA-approved and authorized COVID-19 vaccines. Given the similar course of COVID-19 disease in adults and pediatric patients, today’s approval of Veklury in certain pediatric patients is supported by efficacy results from phase 3 clinical trials in adults. The only approved dosage form is Veklury for injection.
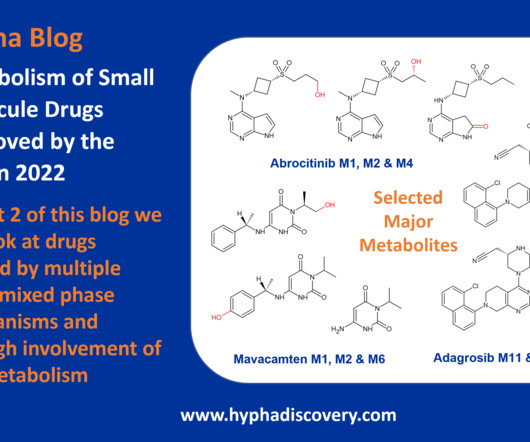
Metabolism of 2022 FDAapproved small molecule drugs part 2 Mixing it Up By Julia Shanu-Wilson In Part 1 of this topic we looked at metabolism of the small molecule drugs approved by the FDA in 2022 that were mediated by CYP3A4. 8 This is not the only point of interest. We hope it was a useful two-parter! Br J Pharmacol.
Its indicated dose of 8 mg is notably higher than the previously approved doses of 2 and 4 mg for other naloxone nasal spray products. . It also provided supporting pharmacokinetic data demonstrating the opioid antagonist’s safety and efficacy. . New indications.
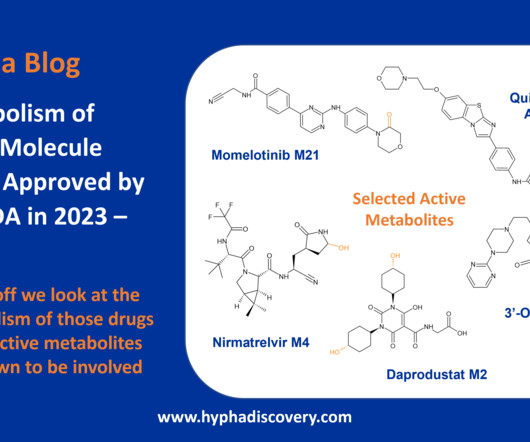
Metabolism of 2023 FDAApproved Small Molecules – PART 1 By Julia Shanu-Wilson 2023 was a fruitful year for drug approvals by the FDA, with a crop of 34 small molecules out of a total of 55 new drugs [1]. link] [10] FDA prescribing information for gepirone. Pharmacokinetics and Metabolism of Nirmatrelvir.
New Indication for Amgen’s Fifth FDA-approved Biosimilar. Now Approved to Treat All Available Rituxan ® Indications. Overall, 311 patients were randomized and treated with RIABNI, rituximab RP approved in the EU (rituximab-EU) or rituximab RP approved in the US (rituximab-US).
A significant effort has been made to correctly map the drug form of the EMA data by manually inspecting different EMA sources of information, such as the Product Information (Annex I: Summary of Product Characteristics and Annex III: Labelling and Package Leaflet) and/or Assessment Report, where available. University of Dundee: T.
The PAS was supported by positive topline data from the REFLECTIONS B538-12 study which evaluated multiple switches between treatment with ABRILADA and its reference product, Humira, both of which were administered with methotrexate in adult patients with moderate to severe rheumatoid arthritis (RA).
.
Depressive Disorders:
Depressive disorders (including depressed mood, depression, major depression, mood altered, mood swings, dysphoria, negative thoughts, suicidal ideation or attempt) have been reported with CABENUVA or the individual products.
US Approval January 2021.
Warnings and Precautions.
This fresh release comes with a few new data soures and also some new features: we added bioactivity data for understudied SLC targets from the RESOLUTE project and included a flag for Natural Products and for Chemical Probes. An an annotation for the ACTION_TYPE of a measurement was included for approx. 270 K bioactivities.
The 505(b)(2) new drug application (NDA) pathway offers a unique opportunity for small molecule developers to bring innovative products to market more efficiently by leveraging existing data they do not own or have right of reference to. The new formulation resulted in a drug product that was very viscous. Human factors.
Even for repurposed drugs being developed under the 505(b)(2) New Drug Application (NDA) pathway, it is critical to review the existing nonclinical and clinical data on the drug to determine what nonclinical studies may be beneficial to conduct prior to the PIND meeting and include this information in the package.
The FDAapproved the first gene therapy in 2017 for certain pediatric and young adult patients with a form of acute lymphoblastic leukemia (ALL), and as of December 2023, there are over 30 approved cell and gene therapies on the market in the U.S.A. Asia, and Europe.
of new cancer drugs tested in Phase I were likely to receive Food and Drug Administration (FDA) approval. years , with biotechnology-derived products potentially adding another year or more. Participants may balk at certain protocol requirements, such as extended visits for pharmacokinetic (PK) measurements.
The review is being conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence (OCE), which provides a framework for concurrent submission and review of oncology products among international partners. Takeda has established an Expanded Access Program (EAP) ( NCT04535557 ) for patients in the U.S.
NYSE American: PLX) (TASE: PLX), a biopharmaceutical company focused on the development, production and commercialization of recombinant therapeutic proteins produced by its proprietary ProCellEx ® plant cell-based protein expression system, and Chiesi Global Rare Diseases, a business unit of Chiesi Farmaceutici S.p.A.,
NYSE American:PLX) (TASE:PLX), a biopharmaceutical company focused on the development, production and commercialization of recombinant therapeutic proteins produced by its proprietary ProCellEx ® plant cell-based protein expression system, and Chiesi Global Rare Diseases , a business unit of Chiesi Farmaceutici S.p.A., Galactosidase-A enzyme.
Food and Drug Administration (“FDA”) for the treatment of agitation associated with delirium. With no FDA-approved treatments for this condition, current guidelines recommend sedative medications to maintain a light level of sedation in adult patients, which is frequently not achieved with commonly used therapies.
Roche’s Chief Medical Officer and Head of Global Product Development. About the FENopta study The FENopta study is a global Phase II, randomised, double-blind, placebo-controlled 12-week study to investigate the efficacy, safety and pharmacokinetics of fenebrutinib in 109 adults aged 18-55 years with RMS.
Until recently, the FDA relied on a monograph process through which firms could bring OTC drugs to market without FDAapproval so long as it adhered to pre-set terms under the monograph. The FDA will follow these procedures for both agency-initiated operations (e.g.,
Additionally, Regeneron bispecifics are manufactured using similar approaches used for human monoclonal antibody medicines, yielding similar properties and pharmacokinetics. These allow for the creation of bispecific antibodies that closely resemble natural human antibodies with no linkers or artificial sequences.
See Warnings and Precautions in the FDA-approved full Prescribing Information for additional information on risks associated with longer-term treatment with baricitinib. It is approved in the U.S. Olumiant was recently approved in Japan for the treatment of pneumonia associated with COVID-19 in hospitalized adult patients.
Evaluation of the Pharmacokinetic Interaction and Safety of Coadministered Atogepant and Topiramate. Optimal Acute Treatment Is Associated With Productivity Gains in People With Migraine: Results From the Chronic Migraine Epidemiology and Outcomes (CaMEO) Study. Abstract Lecture. September 12, 2021. 2:45-2:55 a.m. September 8, 2021.
FDA updates guidance on developing drugs for Covid-19, replacing pandemic-era version Last week, FDA published the third update to its guidance on the development of products to prevent or treat Covid-19. Read Agency IQ’s complete breakdown of the FDA’s PHE guidance documents here.]
Part B of the Phase 3 trial was informed by Part A , an open-label, single-ascending-dose, sequential cohort Phase 2 trial designed to assess the pharmacokinetics and safety of Dupixent in children aged 6 months to 5 years with uncontrolled severe atopic dermatitis.
FDA and global approvals for our groundbreaking therapies in SMA and NMOSD, Roche’s data at AAN reflect our continued commitment to meaningful therapeutic progress for people living with neurological disorders,” said Levi Garraway, M.D., Roche’s Chief Medical Officer and Head of Global Product Development. “We
BLAZE-4 Additionally, initial results from the ongoing BLAZE-4 trial provide viral load and pharmacodynamic/pharmacokinetic data which demonstrated lower doses, including bamlanivimab 700 mg and etesevimab 1400 mg together, are similar to bamlanivimab 2800 mg and etesevimab 2800 mg together. Bamlanivimab FDAApproval History.
BY AMANDA CONTI, ALEXANDER GAFFNEY, MS, RAC SEP 18, 2023 9:24 PM CDT Background: FDA’s standards for evidence When seeking approval from the FDA, companies are required to demonstrate that their product is safe and effective when used as intended. In one landmark case, Warner-Lambert Co.
The Phase I/IIa clinical trial is a randomized, double-blind, placebo controlled, single and multiple dose escalation study to assess the safety, tolerability, pharmacokinetics and pharmacodynamics of NMD670 in 79 male and female healthy subjects and patients with myasthenia gravis. The secondary outcome involves pharmacokinetic endpoints.
FDA-approved oral prescription medicine, 120 mg or 160 mg dependent on weight (<50 kg or ?50 i Continued approval may be contingent upon verification and description of clinical benefit in confirmatory trials. Retevmo may affect both tumor cells and healthy cells, which can result in side effects. Retevmo is an U.S.
Food and Drug Administration (FDA) approval for nmCRPC on February 14, 2018 and was approved for mCSPC on September 17, 2019. In a dedicated drug interaction trial, co-administration of ketoconazole, a strong inhibitor of CYP3A4, had no clinically meaningful effect on the pharmacokinetics of abiraterone.
Read Safety and pharmacokinetics of escalating doses of neutralising monoclonal antibody CAP256V2LS administered with and without VRC07-523LS in HIV-negative women in South Africa (CAPRISA 012B): a phase 1, dose-escalation, randomised controlled trial. (h/t Ricciardi M.J. Science Translational Medicine. ” Paul Sawers. Tech Crunch.
The State of Current Scientific Knowledge Regarding Marijuana HHS found that marijuana’s pharmacokinetic profile varies depending on the route of administration. Basis at 18; NPRM at 44,605. Basis at 24. Basis at 63-64.
The FDAapproved Evrysdi in August 2020 as the first and only at home SMA treatment with proven efficacy in adults, children and infants 2 months and older. Chief Medical Officer and Head of Global Product Development. fold from baseline in the high-dose cohort at 12 months. mg/kg for Part 2. The study met its primary endpoint.
In August, the FDAapproved Evrysdi for the treatment of SMA in adults and children 2 months and older. Roche’s Chief Medical Officer and Head of Global Product Development. Evrysdi is designed to treat SMA by increasing and sustaining the production of the survival of motor neuron (SMN) protein.
Chief Medical Officer and Head of Global Product Development. Evrysdi is a survival of motor neuron 2 (SMN2) splicing modifier designed to treat SMA by increasing and sustaining production of the survival of motor neuron (SMN) protein. Evrysdi is administered daily at home in liquid form by mouth or by feeding tube.
Inhibition of drug transporter breast cancer resistance protein has no effect on the pharmacokinetics of major active metabolites of ozanimod. Food and Drug Administration (FDA) approved Zeposia for the treatment of adults with relapsing forms of multiple sclerosis (RMS) in March 2020. FDA-APPROVED INDICATION FOR ZEPOSIA.
Limited Evidence for Nalfmefene "In 2021, due to the widespread availability of high-potency synthetic opioids like fentanyl, the US FDAapproved two high-dose naloxone products, an 8 mg IN spray (Kloxxado) and a 5 mg IM injectable (Zimhi). In 2023, the FDAapproved a 2.7 In 2023, the FDAapproved a 2.7
The suspension was deemed too thick and therefore unsuitable for production. 1] Society and culture Legal status Benzgalantamine was approved for medical use in the United States in July 2024. [1] 1] Society and culture Legal status Benzgalantamine was approved for medical use in the United States in July 2024. [1] 29 July 2024.
9] Tiredness, febrile neutropenia (low white blood cell counts with fever) and high blood levels of bilirubin (a breakdown product of red blood cells) are also seen in more than 1 in 10 adults, and rash also affects more than 1 in 10 children. [9] TREOSULFAN C 6 H 14 O 8 S 2 MW 278.29 9] Efficacy was evaluated in MC-FludT.14/L
Zidovudine showed promise against multiple HIV strains in cultured cells, and the Food and Drug Administration (FDA) approved it for human studies within five months. By 1987, the FDA licensed zidovudine after trials showed it increased survival rates.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.






































Let's personalize your content