This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Food and Drug Administration (FDA) plays a pivotal role in fostering the development of treatments for rare diseases through its Orphan Products Grants Program. Each year, FDA selects a limited number of clinical trials to fund to help sponsors pursue development of medical products for rare diseases and advance their field.
In order to understand the current status on cannabis and CBD research studies, it’s important to understand where each product stands. Cannabis and cannabis-derived compounds are treated the same as other FDA-regulatedproducts and subject to the same authorities and requirements. The 2018 Farm Bill.
Food and Drug Administration approved the first interchangeable biosimilar insulin product, indicated to improve glycemic control in adults and pediatric patients with Type 1 diabetes mellitus and in adults with Type 2 diabetes mellitus. for the treatment of diabetes. Biosimilars marketed in the U.S.
Genentech’s once-daily oral therapy Gavreto (pralsetinib) has secured FDA backing in the treatment of metastatic rearranged during transfection (RET) fusion-positive non-small cell lung cancer (NSCLC), it has emerged. The decision was made under accelerated approval, marking Genentech’s sixth approval from the US regulator within lung cancer.
FDAApproves Danyelza (naxitamab-gqgk) for the Treatment of Neuroblastoma. the “Company” or “Y-mAbs”) (Nasdaq: YMAB) a commercial-stage biopharmaceutical company focused on the development and commercialization of novel, antibody-based therapeutic products for the treatment of cancer, today announced that the U.S. NEW YORK, Nov.
NS Pharma has claimed a tentative FDAapproval for its Viltepso (viltolarsen) injection in the treatment of Duchenne muscular dystrophy (DMD) in patients who are suitable to receive exon 53 skipping therapy. Based on these data, the FDA judged that Viltepso is “reasonably likely” to provide clinical benefit in the approved indication.
Valentine — On November 22, 2022, FDAapproved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
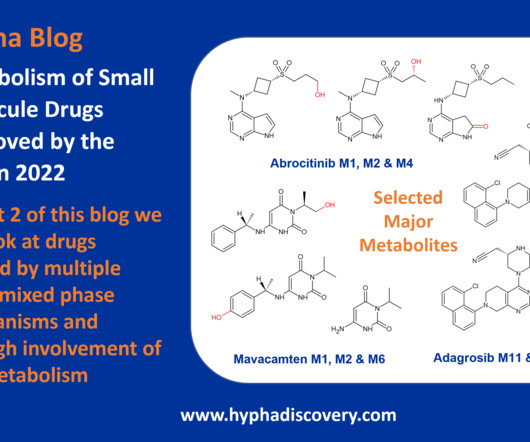
Metabolism of 2022 FDAapproved small molecule drugs part 2 Mixing it Up By Julia Shanu-Wilson In Part 1 of this topic we looked at metabolism of the small molecule drugs approved by the FDA in 2022 that were mediated by CYP3A4. 8 This is not the only point of interest. We hope it was a useful two-parter!
FDAApproves Klisyri (tirbanibulin) for the Treatment of Actinic Keratosis on the Face or Scalp. Klisyri is the first FDAapproved branded proprietary product for Athenex and will be launched in partnership with Almirall in the U.S. The FDAapproval of Klisyri is a significant milestone for Athenex.
As we explained back in March , Teva had initiated Hatch-Waxman pre-launch patent litigation against Amneal for infringement of 5 Orange Book-listed patents reading on the device constituent (a metered dose inhaler) of Teva’s combination product ProAir HFA. b)(1) that drug product patents may be listed if they claim the “drug product.
Food and Drug Administration (FDA) has approved commercial production at the company’s new CAR T-cell therapy manufacturing facility in Frederick, Maryland. The site will produce Kite’s FDAapproved CAR T-cell therapy used to treat blood cancer. Kite, a Gilead Company (Nasdaq: GILD), today announced the U.S.
The Act is intended to address national security concerns by prohibiting certain conduct by regulated industry. Effect on FDAapprovals— In cases where a biological product or drug needs to change aspects of its manufacturing processes to avoid using a covered equipment or service, will it need to file supplements with FDA for the CMC update?
Start Up and Generic Pharmaceutical Drug and Biologic Companies have high quality, affordable products and biosimilars that improve the quality of life for their patients. has over 30 years of management, full spectrum Regulated Life Sciences, RA, QA, EU-MDR, QMS, PMS, CSV/CSA, and R&D experience. Author Information William E.
Start Up and Generic Pharmaceutical Drug and Biologic Companies have high quality, affordable products that improve the quality of life for their patients. In a FDA Pre-Submission that leads to FDAApproval, more does not equal better and more does not equal relevance to a specific Pharmaceutical Drug or Biologic Product.
FDA finalizes guidance on electronic submissions for OTC productsFDA has fulfilled its commitment under the Over-the-Counter Monograph Drug User Fee (OMUFA) program to issue final guidance on how sponsors can electronically submit monographs and other documents. Read AgencyIQ’s explainer on the CARES Act here.
This activity may be regulated by the K-loop, which is known to be important for KIF1A’s long-range movement. Taking a drug that shelved during development or didn’t receive FDAapproval, and assessing its application for another disease, is called drug repositioning.
Won will share his expertise and insights on the intricacies of the United States Food and Drug Administration (FDA) regulations pertaining to medical devices, with a special focus on class II and class III hearing devices. The comprehensive agenda for the symposium can be accessed in its entirety here.
regulators to seek approval of our COVID-19 vaccine based on our pivotal Phase 3 trial and follow-up data.”. Food and Drug Administration (FDA), but has been authorized for emergency use by FDA under an Emergency Use Authorization (EUA) to prevent Coronavirus Disease 2019 (COVID-19) for use in individuals 16 years of age and older.
It involves designing, analyzing, and controlling manufacturing by taking timely measurements to ensure final product quality, efficacy, and safety. Pharmaceutical Product Development is an inventive insertion in pharmaceutical industries and its regulatory authorities. So design quality is linked to product performance.
The FTC expressed concern that patent listings that do not meet the statutory criteria undermine the competitive process, may disincentivize investment in developing generic and follow-on products, and reduce patient access to more affordable drugs thereby increasing costs to the healthcare system.
The American Conference Institute (“ACI”) will be hosting the go-to forum for critical updates on OTC regulation and enforcement, monograph reform, ACNU and advertising essentials… and FDA Law Blog readers can get a discount. Deb along with fellow panelists Kyle Y.
156, a patent may be extended only once (even if it would be eligible for extension on more than one occasion because it applies to several FDA-approvedproducts), and only one patent may be extended for each regulatory review period. FDA-2020-E-1840 (July 13, 2020). To that end, 35 U.S.C. § 156(c)(4) 351, 355-56 (1985).
By Riëtte van Laack — For those readers unfamiliar with the regulation of animal food ingredients in the United States, below is a brief background. In the United States, animal food (feed) regulations are enforced by state and federal regulatory officials. States frequently review labels (and labeling) for animal food products.
For purposes of determining the date on which a product receives permission under the second sentence of this paragraph, if such permission is transmitted after 4:30 P.M., Eastern Time, on a business day, or is transmitted on a day that is not a business day, the product shall be deemed to receive such permission on the next business day.
Now, the FDA has released guidance laying out the tests and regulatory submissions needed for the reformulation of products containing these ingredients. Benzene may occur in some products from the degradation of other ingredients, increasing the risk for unintentional contamination.
Koblitz — In the world of patent term extensions, every day considered part of the regulatory review period is important, as that day—either in whole or in part—gets added back to the patent upon approval of the product. The “review phase” is the period between the initial submission and approval of the NADA.
Amongst other things, FDA co-opted many of the same definitions for key terms for implementation of the BPCIA. FDA borrowed this definition from 21 C.F.R. mg/mL) for liquid parenteral drug products. mg/mL) for liquid parenteral drug products. FDA explained that its bioequivalence regulations at 21 C.F.R.
Food and Drug Administration (FDA) has approved the company’s supplemental Biologics License Application for Xolair® (omalizumab) prefilled syringe for self-injection across all approved U.S. Roche’s Chief Medical Officer and Head of Global Product Development. with Xolair since its initial approval in 2003.
The personalised cancer treatment has received conditional approval in the US.
US regulators have approved Genentech’s Gavreto (pralsetinib) for the treatment of adults with metastatic rearranged during transfection (RET) fusion-positive non-small cell lung cancer (NSCLC).
Pfizer plans to file for full FDAapproval of Covid vaccine at the end of this month ( CNBC ).
The FDA is set to authorize the Pfizer-BioNTech vaccine for those 12-15 years old by early next week.
Big three drug distributors blame doctors, regulators in trial over opioid epidemic ( Reuters ).
We blogged about CVM last week and explained the increasing attention to animal health products due to the expansion of the animal and pet product market. District Court challenging FDA’s plan to remove their products from the market. Specifically, FDA is looking to remove a drug called carbadox from the market.
Food and Drug Administration (FDA)-approved therapies for treating PAH were primarily vasodilators, designed to overcome the imbalance between vasoactive and vasodilator mediators and to restore endothelial cell function. Historically, the available drugs and U.S.
The collaboration with Bruker announced today will assess the suitability of the test as a professional-use in-vitro diagnostic (IVD) product for SARS-CoV-2 infection to run on Bruker’s MALDI-TOF instruments for sale in the UK and Europe. I look forward to updating the market when we have definitive clinical performance data.”.
Late last week, the FTC submitted an Amicus Brief in recent Hatch-Waxman patent litigation in the District Court of New Jersey between Reference Listed Drug holder Teva Branded Pharmaceutical Products R&D Inc. Because a “drug product” is defined by regulation as a “Finished dosage form.
Specifically, FDA determined that Avadel’s Lumryz can “break” Jazz’s ODE because it is “clinically superior” based on Lumryz’s “major contribution to patient care,” rendering it not “the same drug” under the Orphan Drug Act. 360cc(c), the statute does not permit FDA to promulgate regulations to use clinical superiority to break ODE.
Food and Drug Administration has scheduled a meeting of its Vaccines and Related Biological Products Advisory Committee (VRBPAC) on Dec. The FDA recognizes that transparency and dialogue are critical for the public to have confidence in COVID-19 vaccines. Source: FDA. BNT162b2 (SARS-CoV-2 vaccine) FDAApproval History.
Under section §812.10, a sponsor may request a waiver of any requirement of the IDE regulations through an application with supporting documentation. The FDA may grant a waiver if the requirement is not stipulated by the Federal Food, Drug, and Cosmetic (FD&C Act) or if it is not necessary for the protection of human subjects. [3]
In 2016, the Food and Drug Administration (FDA) approved Spinraza (nusinersen). While the FDA’sapproval of nusinersen may not seem extraordinary, it was. Nusinersen’s approval marked the first time nonclinical data supported conducting initial clinical trials involving children. Why This Guidance Now?
This feedback prompted the FDA to release a second paper in 2021 titled “ Artificial Intelligence/Machine Learning (AI.ML)-Based Software as a Medical Device (SaMD) Action Plan ,” which outlined the agency’s five-pillar approach to regulating AI/ML. AI can help monitor product quality and detect deviations.
156, as added by the 1984 Hatch-Waxman Amendments, for certain FDA-regulatedproducts. The use of a surrogate endpoint can considerably shorten the time required prior to receiving FDAapproval. Karst — Earlier this week, we posted Part 1 of our three-part series on U.S.
Our team can help to determine if obtaining the EUA is an option for your medical device product. Post EUA documents: EUA issuance does not imply approval or approval from the FDA. This product is permitted only in a public health emergency when the EUA is no longer in effect. What is meant by CGMP?
Koblitz — After years of silence from FDA on whether certain patents could be listed in the Orange Book, some manufacturers of drug and device combination products have had a rude awakening lately. In fact, federal law and regulation appear to require AbbVie to list these patents.”
1 In 2017, the US Food and Drug Administration (FDA) approved the first AAV-based gene replacement therapy (Luxturna), for Leber congenital amaurosis type 2. 2 Since then, the FDA has approved four more AAV-based gene therapies—Zolgensma, Hemgenix, Elevidys and Rocktavian—for treating various diseases.
FDA-2023-P-0313 and FDA-2023-P-0344 ) regarding its product Hetlioz (tasimelteon). Vanda requested that FDA revoke the approval of Apotex’s and Teva’s generic versions of Hetlioz on the grounds that the generic tasimelteon products did not meet the statutory “same labeling” requirement for generic drugs found in 21 U.S.C. §
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.













































Let's personalize your content