This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Additionally, the immunogenicity of biosimilars, which is the ability of the drug to trigger an immuneresponse, can be highly variable and impact the safety and efficacy of the drug.
Researchers must characterize the anti-drug-antibody (ADA) response in preclinical and clinical studies and report any ADA-positive samples as a risk-based approach. These interactions may disrupt protein function, trigger immuneresponses, or contribute to other toxicological risks.
Also recently, FDA’s Cellular, Tissue, and Gene Therapies Advisory Committe turned down a stem cell treatment for amyotrophic lateral sclerosis, aka ALS, Lou Gehrig’s disease, or motor neuron disease. If MSCs come from the patient (autologous), then an immuneresponse shouldn’t be a hindrance.
This is particularly pertinent in immune-based cell therapies like CAR-T, where animals do not mimic the human immuneresponse. This was recognised in the FDA Modernization Act 2.0, In orphan and rare diseases, it is hard to get an animal model to represent a disease state that is poorly understood.
Food and Drug Administration (FDA) has granted Fast Track Designation for NVX-CoV2373, the Company’s COVID-19 vaccine candidate. Both vaccine candidates incorporate Novavax’ proprietary saponin-based Matrix-M adjuvant to enhance the immuneresponse and stimulate high levels of neutralizing antibodies.?Novavax?is Source link.
Food and Drug Administration (FDA) to support the evaluation of a third, or booster, dose of the companies’ COVID-19 vaccine (BNT162b2) for future licensure. “The We are pleased to submit these data to the FDA as we continue working together to address the evolving challenges of this pandemic.”. “We CEO and Co-founder of BioNTech.
Food and Drug Administration (FDA) has approved EYSUVIS for the short-term treatment of dry eye disease. . EYSUVIS is the first FDA-approved corticosteroid specifically for dry eye disease treatment. The FDA approved EYSUVIS based on the positive results from one Phase II and three Phase III trials. Roughly 16.4 million U.S.
Nasdaq: MBRX) (Moleculin or the Company), a clinical stage pharmaceutical company with a broad portfolio of drug candidates targeting highly resistant tumors and viruses, today announced that the US Food and Drug Administration (FDA) has granted Orphan Drug Designation (ODD) to Annamycin for treatment of soft tissue sarcomas. .
In a Phase 2/3 clinical trial, the Pfizer-BioNTech COVID-19 Vaccine elicited a strong immuneresponse in this age group Three 3-µg doses had favorable safety profile similar to placebo in young children ages 6 months through 4 years in Phase 2/3 clinical trial Pfizer-BioNTech COVID-19 Vaccine now authorized in the U.S. Pfizer Inc.
The cocktail reduced viral load “with a greater effect in patients whose immuneresponse had not yet been initiated or who had a high viral load at baseline,” the researchers said. The FDA granted Regeneron’s antibody cocktail Emergency Use Authorization in November.
Our results show that the candidate vaccine formulation is safe, produces rapid immuneresponses – within seven days – and elicits comprehensive immunity against SARS-CoV-2,” said Varadarajan. A fundamental limitation of intramuscular vaccines is that they’re not designed to elicit mucosal immunity.
Food and Drug Administration (FDA) has cleared the Company’s Investigational New Drug (IND) application for ADI-001, an allogeneic gamma delta (??) The clearance of the IND for ADI-001 by the FDA is a significant milestone in the development of CAR ?? T cell therapy in NHL patients. Forward-Looking Statements.
The Weinreich article also reports that the “antibody cocktail reduced viral load, with a greater effect in patients whose immuneresponse had not yet been initiated or who had a high viral load at baseline. 24, 2020 ). Safety outcomes were similar in the combined REGN-COV2 dose groups and the placebo group.”.
Secondary objectives are to describe immuneresponses produced by each of the vaccines. Food and Drug Administration (FDA), but has been authorized for emergency use by FDA under an Emergency Use Authorization (EUA) to prevent Coronavirus Disease 2019 (COVID-19) for use in individuals 12 years of age and older.
under an Emergency Use Authorization (EUA) granted by the FDA on December 11, 2020. Submission of a BLA, which requires longer-term follow-up data for acceptance and approval, is the next step in the rigorous FDA review process. “We The Pfizer-BioNTech COVID-19 Vaccine is currently available in the U.S.
.” The clinical trial involved 24 COVID patients at one of two Miami-area hospitals who had developed severe acute respiratory distress syndrome, a condition in which the body’s immuneresponse to a serious infection causes the lungs to fill with fluid.
Strong Th1 cell-mediated immuneresponses were also observed for the vaccine candidates with either adjuvant. We are encouraged by the high level of neutralizing antibodies in combination with the strong Th1 response which we believe could play an important role in controlling infection.
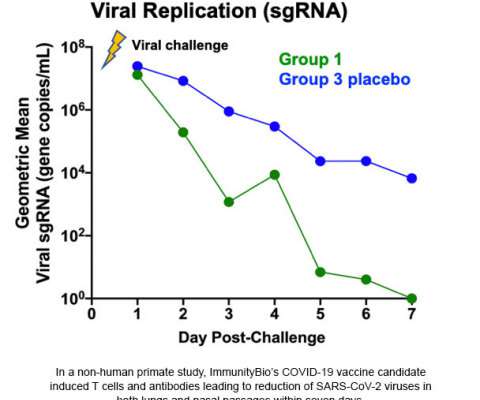
The vaccine targeted both the inner nucleocapsid (N) and the outer spike (S) proteins of the virus to maximize the immuneresponse. The study showed this broad immuneresponse led to the complete clearance of the virus in a matter of days after infection of previously-vaccinated primates.
3 Additionally, iptacopan has the benefit of targeting factor B, which only affect the alternative complement pathway, leaving the classic and lectin pathway untouched for the body to still mount adequate immuneresponses against pathogens. Food and Drug Administration (FDA). 1] It is taken by mouth. [1] 5 December 2023.
Draft guidance on potency assays for CGT products garners extensive stakeholder input Late last year, the FDA published a draft update to its 2011 guidance on potency assays for cell and gene therapy products, unveiling a major shift in approach to the issue. This is a question that sponsors have been struggling with for quite some time.
Preliminary blinded data on NVX-CoV2373 in older adults needed to proceed to Phase 3 has previously been positively reviewed by the Food and Drug Administration (FDA). Both vaccine candidates incorporate Novavax’ proprietary saponin-based Matrix-M adjuvant to enhance the immuneresponse and stimulate high levels of neutralizing antibodies.?Novavax?is
Food and Drug Administration (FDA) gave the go-ahead for the company to initiate a Phase I trial of PRGN-2012 in adults with recurrent RRP. The rest are in Phase I trials for ovarian cancer, AML, MDS, heart failure, HPV+ solid tumors and recurrent respiratory papillomatosis (RRP). The company also has an extensive preclinical pipeline.
The FDA is expected to assemble its vaccines advisory committee Dec. Former FDA Commissioner Scott Gottlieb hailed Pfizer/BioNTech’s success as a “game change” and predicted their vaccine could be the solution to the health crisis. 8-10 to discuss the recent developments, including Pfizer/BioNTech’s and Moderna’s candidates.
While its involvement in the do-not-eat-me signal from cancer has inspired therapeutic development of this pathway for oncology, the function of the innate immune checkpoint we identified in 2009 1 extends to both innate and adaptive immuneresponses. CD24 and Siglec-10 Selectively Repress Tissue Damage-Induced ImmuneResponses.
The Committee considered that the available evidence was sufficient to conclude that the immuneresponse to a booster dose in adolescents would be at least equal to that in adults. Food and Drug Administration (FDA) earlier this year as an expansion to the existing EUA for the primary series.
BTK is an intracellular signaling molecule involved in innate and adaptive immuneresponses involved in certain immune-mediated diseases. Rilzabrutinib was granted FDA Fast Track Designation for ITP in November 2020 and for pemphigus vulgaris in May 2021.
This is due to the high-risk nature of these kinds of products (especially for a first-in-class therapy) and the fact that some cell or gene therapies can only be administered once due to the development of immuneresponses to the drug product.
Food and Drug Administration (FDA) and the Moderna vaccine is soon to follow, here’s a look at several of the top COVID-19 vaccine candidates and where they stand as of today. The Pfizer – BioNTech COVID-19 vaccine was sent to the FDA for possible Emergency Use Authorization (EUA) on Friday, November 20. Also, the U.S.
Adjuvanted S-Trimer COVID-19 vaccine candidates demonstrated favorable safety and tolerability profiles and strong neutralizing immuneresponses in a phase 1 trial.
FDA approval for prevention of infection caused by all known subtypes of hepatitis B virus in adults age 18 years and older.
CHENGDU, China , Feb.
Food and Drug Administration (FDA), but has been authorized for emergency use by FDA under an Emergency Use Authorization (EUA) to prevent Coronavirus Disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) for use in individuals 12 years of age and older. .:
On January 12, its VITROS SARS-CoV-2 Antigen Test for mass-scale COVID-19 testing received FDA Emergency Use Authorization. The company is working on a new approach to immunotherapy that uses the body’s own T-cells to create specific, potent, and durable immuneresponses for cancer and other life-threatening immune-mediated disease.
In late October, SetPoint Medical received Breakthrough Device Designation from the FDA for the use of its novel bioelectronic device by patients with rheumatoid arthritis (RA) who have incomplete response to, or are intolerant to multiple biologic drugs. The FDA has set an action date of July 7, 2021. Bioelectronic Platform.
The study assesses the safety profile and immuneresponse when COVID-19 mRNA investigational booster vaccine (100 mcg dose) and high-dose quadrivalent influenza vaccine are administered simultaneously. Full results of the study will be published later this year. About Fluzone ® High-Dose Quadrivalent / Efluelda ®.
An expert review by a world group of scientists, including some at the WHO and FDA, concludes that, even for the delta variant, vaccine efficacy against severe COVID is so high that booster doses for the overall population aren’t appropriate at this stage within the pandemic. Considerations in boosting COVID-19 vaccine immuneresponses.
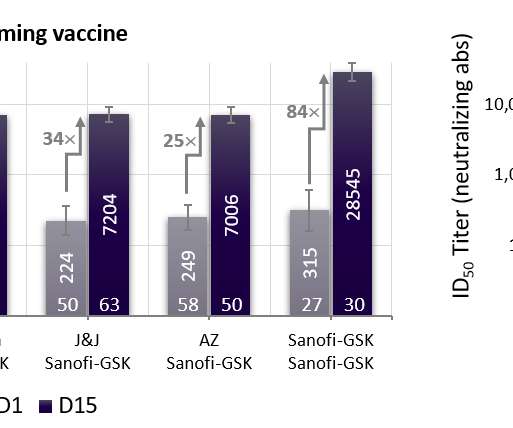
The public health relevance of the refrigerator temperature-stable adjuvanted protein-based Sanofi-GSK vaccine is strongly supported by the induction of robust immuneresponses and a favorable safety profile in multiple settings. Figure 1a – Pre- vs post-booster neutralizing antibody titers in 18-55-yr old participants.
The Phase 1 interim analysis showed that mRNA-1273 was generally well-tolerated across all age groups and induced rapid and strong immuneresponses against SARS-CoV-2. mRNA-1273 is currently being studied in a Phase 3 randomized, 1:1 placebo-controlled trial of 30,000 participants at the 100 µg dose level in the U.S.
Food and Drug Administration (FDA), but has been authorized for emergency use by FDA under an Emergency Use Authorization (EUA) to prevent Coronavirus Disease 2019 (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) for use in individuals 12 years of age and older. .: BioNTech COVID-19 Vaccine.
1 In 2017, the US Food and Drug Administration (FDA) approved the first AAV-based gene replacement therapy (Luxturna), for Leber congenital amaurosis type 2. 2 Since then, the FDA has approved four more AAV-based gene therapies—Zolgensma, Hemgenix, Elevidys and Rocktavian—for treating various diseases.
Clover’s S-Trimer antigen adjuvanted with CpG 1018 plus alum demonstrated low reactogenicity while providing high levels of neutralizing antibodies and a strong Th1-biased cell-mediated immuneresponse. Food and Drug Administration (FDA).
The FDA’s approval of the Pfizer-BioNTech vaccine, for example, listed pain at the injection site, tiredness, headache, muscle pain, chills, joint pain and fever as common reactions.
FDA authorizes COVID-19 mRNA vaccine for emergency use; companies are prepared to deliver first doses in the U.S. Food and Drug Administration (FDA) has authorized the emergency use of the mRNA vaccine, BNT162b2, against COVID-19 in individuals 16 years of age or older. Friday, December 11, 2020.
immediately.
(Nasdaq: TNXP) (Tonix or the Company), a clinical-stage biopharmaceutical company, today announced that the first participant was enrolled in the observational PRECISION study (TNX-C002), to examine the immuneresponses to COVID-19 in healthy volunteers who have recovered from COVID-19 or were asymptomatic.
While further study is needed to understand all the details and their implications, it suggests that this interaction may alter important aspects of the human immuneresponse, including blocking interferon signals that are crucial for sounding the alarm to prevent serious illness.
Decreased levels of PGRN, a key regulator of immuneresponse, lysosomal function, and neuronal survival in the brain, are genetically linked to many neurodegenerative disorders. There are currently no FDA-approved treatment options for FTD. About the Progranulin-Elevating Monoclonal Antibodies – AL001 and AL101.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.





































Let's personalize your content