This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
This article was originally published by Ioana Gherghescu and Begoña Delgado-Charro in Pharmaceutics 2021, 13(1) under a Creative Commons Attribution License. The post The Biosimilar Landscape: An Overview of Regulatory Approvals by the EMA and FDA appeared first on DrugPatentWatch - Make Better Decisions.
Elsewhere, former NCI director Ned Sharpless founded a new startup and Novartis licensed another radiopharma drug. The agency turned back Astellas’ attempt to update its drug Izervay’s labeling.
The biological license application (BLA) is one of the many requests for marketing approval received by the FDA. Unlike the New Drug Application (NDA) which is usually the go-to submission for chemically synthesized, low molecular weight drugs BLAs grant sponsors the ability to introduce Biologics into interstate commerce.
FDA once estimated that OTC hearing aids would save patients over $3000.) The category of OTC hearing aids was created by congressional mandate and implemented by FDA to address an “unmet public health need.” This is because “FDA officials and six external stakeholder groups. .
Who better than people living with a condition to inform drug companies, physicians, academics, and the FDA on what it is like to live with their condition, what symptoms most impact their lives, what goes into their decision about whether to participate in a clinical trial, and what kind of treatment effects would be most meaningful to them?
Karst — While the Biologics Price Competition and Innovation Act (“BPCIA”) is inherently distinct from the Hatch-Waxman Act, many of the fundamental concepts FDA adopted as it enacted the Hatch-Waxman Act made their way into FDA’s implementation of the BPCIA. FDA borrowed this definition from 21 C.F.R.
Valentine — On November 22, 2022, FDA approved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
s (Bridge) opioid addiction development therapeutic product, BT-219, and executed an Exclusive Licensing Agreement to use Catalent’s proprietary Zydis® orally disintegrating tablet (ODT) technology. FDA for BT-219 under the “505(b)(2)” regulatory submission pathway as well as a possible future single entity buprenorphine product.
NYSE: PFE) and BioNTech SE (Nasdaq: BNTX) today announced they have submitted a supplemental Biologics License Application (sBLA) to the U.S. Food and Drug Administration (FDA) to expand the approval of COMIRNATY® (COVID-19 Vaccine, mRNA) to include individuals ages 12 through 15 years. Pfizer Inc.
HP&M has been helping clients navigate the challenges of state licensing regulations for drugs, biologics, medical devices, OTCs, 503B outsourcing facilities, 503A pharmacies, foods, dietary supplements, cannabis, and wholesalers/distributors for many years. Keup has joined the firm to assist attorneys Karla L.
Houck — In August 2023 the Food and Drug Administration (“FDA”) and Health and Human Services (“HHS”) recommended that the Drug Enforcement Administration (“DEA”) reschedule marijuana from schedule I under the federal Controlled Substances Act (“CSA”) to schedule III. HHS forwards FDA’s analysis and recommendation to DEA. By Larry K.
Gibbs — For more than three decades, FDA has claimed that the Federal Food, Drug & Cosmetic (FD&C Act) gives the agency legal authority to regulate laboratory developed tests (LDTs) as medical devices (see our prior post here ). Javitt & Michael D. Shumsky & Philip Won & Adrienne R. Gaulkin & Jeffrey N.
Mullen — With comments due on the proposed LDT rule last week, FDA wasted no time updating the Unified Agenda to indicate that a final rule may be published in April ( here ). Under the Administrative Procedure Act, FDA is obligated to address major substantive issues when – and not so much if – it publishes a final rule.
Food and Drug Administration (FDA) on Friday, December 11, 2020. During the meeting, the FDA provided encouraging feedback regarding the Phase 3 study of omidubicel pertaining to the pre-specified primary and secondary endpoints. chief executive officer of Gamida Cell.
The Countering Emerging Threats - Rapid Acquisition and Investigation of Drugs for Repurposing (CET RAIDR) program within the JPM Medical is designed to rapidly tackle known, unknown, and emerging threats by utilizing late-stage or licensed therapeutics. Repurposing is one such method.
FDA this year. “Rolontis,” a treatment for neutropenia that had its technology licensed out to Spectrum Pharmaceuticals, Inc. and “Oraxol,” which was licensed out to Athenex, Inc. “Rolontis,” a treatment for neutropenia that had its technology licensed out to Spectrum Pharmaceuticals, Inc.
What We Expect the FDA to do in June 2023 In this ongoing feature, AgencyIQ looks at public data to determine what the FDA is likely to do in the month ahead, including key deadlines, meetings, events, planned regulations, comment periods and more. Fill out the form to read the full article.
Claud — There is a growing consensus among legal experts that after Loper Bright, FDA may rely on non-binding guidance to instruct industry with hopes of charting regulatory pathways that avoid litigation. The top line from FDA for any cGMP-governed industry like these is always going to be that quality matters.
Livornese — On February 6, 2024, FDA issued a draft guidance titled Notifying FDA of a Discontinuance or Interruption in Manufacturing of Finished Products or Active Pharmaceutical Ingredients Under Section 506C of the FD&C Act. By Véronique Li, Senior Medical Device Regulation Expert & Deborah L.
FDA Approves Sesquient (fosphenytoin sodium) for the Treatment of Status Epilepticus in Adult and Pediatric Patients. Food and Drug Administration (FDA) has approved Sesquient (fosphenytoin sodium for injection) for the treatment of status epilepticus in adult and pediatric patients. About Sesquient.
Food and Drug Administration (FDA) announced its acceptance of the Biologics License Application (BLA) for exa-cel. In recognition of the unmet need and medical urgency for innovative therapies in the sickle cell space, the FDA granted exa-cel Priority Review, with a formal decision expected by December 8, 2023.
Food and Drug Administration (FDA) ’s focus appears to be on Emergency Use Authorizations (EUAs) for the Pfizer-BioNTech and Moderna COVID-19 vaccines, as the year wraps up there are still some PDUFA dates on the agency’s calendar. We look forward to working with the FDA to bring ABP 798 to market.”. Although much of the U.S.
The FDA's Biologics License Application (BLA) approvals in 2023 have marked a significant chapter in medical innovation, embodying precision and transformative therapies. A closer examination of the approvals, their numbers, and the trends they reveal unveils a dynamic landscape in biologics.
Food and Drug Administration (FDA) is plenty busy with COVID-19 vaccine Emergency Use Authorizations (EUAs) this month, but they’re also wrapping up the year with a few PDUFA dates for other therapies. Vibegron could be a potentially important and differentiated new oral treatment, if approved by the FDA, for patients suffering with OAB.”.
FDA advisory committees recommended just 50 percent of the 18 new therapies and indications they reviewed in 2020, the lowest rate since 2007, and the agency seems to be reserving the panels for more problematic applications, according to Prevision Policy, a Washington, D.C.-based based research firm.
Food and Drug Administration (FDA) are starting to pick up. People living with LN are in need of an advanced therapy that quickly drives the disease into remission and mediates kidney damage,” said Peter Greenleaf, president and chief executive officer of Aurinia, in July 2020 when the FDA accepted the NDA to review. “We Here’s a look.
Food and Drug Administration (FDA) has a busy end of November planned, with numerous PDUFA dates to address. The FDA approved it under the brand name Gavreto on September 4. Here’s a look at the upcoming week. Blueprint Medicines’ Pralsetinib for Non-Small Cell Lung Cancer. It holds the rights for the drug in the rest of the world.
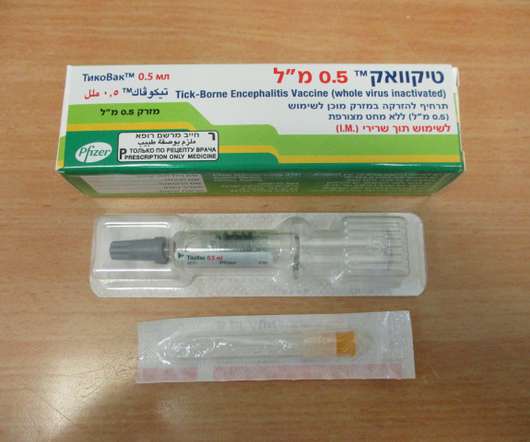
Food and Drug Administration (FDA) has approved TICOVAC (tick-borne encephalitis (TBE) vaccine) for active immunization to prevent TBE in individuals 1 year of age and older. 1 TICOVAC is the only FDA-approved vaccine to help protect U.S. Following today’s FDA approval, the U.S.
New FDA guidance on addressing misinformation under White House review A new question-and-answer draft guidance document focused on “addressing misinformation” about regulated life sciences products is currently under administrative review at the White House’s Office of Information and Regulatory Affairs (OIRA).
FDA draft guidance on addressing misinformation clears White House review A few months ahead of a potential administration transition, the White House has cleared (with changes) a new question-and-answer draft guidance document focused on “addressing misinformation” about regulated life sciences products.
Food and Drug Administration (FDA). Y-mAbs Therapeutics has a target action date of November 30 for its Biologics License Application (BLA) for Danyelza (naxitamab) for patients with relapsed/refractory high-risk neuroblastoma. The FDA approved the drug on November 24. Here’s a look.
FDA Approves Danyelza (naxitamab-gqgk) for the Treatment of Neuroblastoma. Food and Drug Administration (“FDA”) has approved Danyelza (naxitamab-gqgk) 40mg/10ml. The product has received Priority Review, Orphan Drug, Breakthrough Therapy, and Rare Pediatric Disease designations from the FDA. NEW YORK, Nov.
All clinical trial or marketing applications submitted to the FDA must include a form that summarizes the content of the submission and any relevant information on the sponsor and drug for the reviewers. Periodically, the FDA updates these forms to enhance their usability and incorporate relevant content to help sponsors and reviewers.
The test is performed by a qualified, trained operator under the supervision of a health care provider licensed or authorized by state law to prescribe tests and can provide results in less than three minutes. director of the FDA’s Center for Devices and Radiological Health. The FDA, an agency within the U.S.
Food and Drug Administration (FDA) has accepted for review the Prior Approval Supplement (PAS) to the Biologics License Application (BLA) for ABRILADA™ (adalimumab-afzb) as an interchangeable biosimilar to Humira® (adalimumab). The Biosimilar User Fee Act (BsUFA) goal date for an FDA decision is in Q4 2022. “An
FDA ACCEPTS FOR PRIORITY REVIEW PFIZER’S APPLICATION FOR TICOVAC (TICK-BORNE ENCEPHALITIS VACCINE). In line with Priority Review designation, the FDA will target an action within six months of the application submission date, 3 with the anticipated Prescription Drug User Fee Act (PDUFA) action date expected for August 2021.
FDA interprets the exclusionary clause to mean that the critical date is the date that the IND came into effect, not the date on which the existence of the substantial clinical investigations under the IND has been made public. What led to NPA’s lawsuit? The facts are detailed in the complaint.
Many of our team at the FDA are parents and grandparents themselves, and our team shares an equivalent concerns as many in our country about protecting our loved ones from COVID-19. The FDA will work closely with each manufacturer to make sure this data analysis is strong and meets regulatory standards.
Third time’s the charm as Heron wins FDA nod for non-opioid anesthetic Zynrelef ( Endpoints ).
Amgen, AstraZeneca bolster their case for breakthrough asthma program as FDA considers taking up a review ( Endpoints ).
Biogen licenses a stroke drug from Japanese drugmaker TMS ( BioPharmaDive ).
Enter the FDA’s Center for Veterinary Medicine (“CVM”). A longstanding component of the FDA and its predecessor, CVM, in its initial form as part of the Department of Health, Education, and Welfare, was established in 1965 and evolved in CVM by 1984.
While traditionally conducted during Phase 2 or later, the FDA has recently been requesting data sooner in the development process, making it critical to implement proactive AME studies to lay the groundwork for advanced phases of your clinical program. To accomplish this, we pay careful attention to both dosing and sample collection.
Food and Drug Administration (FDA) has cleared an investigational new drug (IND) application for the company’s lead product candidate, PBGM01, an adeno-associated virus (AAV)-delivery gene therapy that is being studied for the treatment of infantile GM1 gangliosidosis (GM1). president and chief executive officer of Passage Bio.
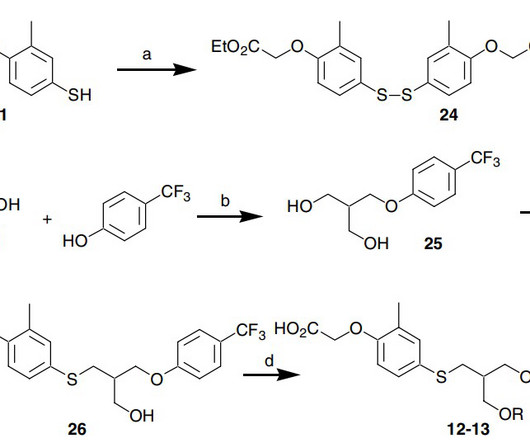
1] [2] [3] The compound was licensed from Janssen Pharmaceutica NV. [4] 1] [2] [3] The compound was licensed from Janssen Pharmaceutica NV. [4] 1] [2] [3] The compound was licensed from Janssen Pharmaceutica NV. [4] FDA” (Press release). Seladelpar cas 851528-79-5 C 21 H 23 F 3 O 5 S, 444.47 doi: 10.1016/j.bmcl.2007.05.007.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.















































Let's personalize your content