This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Valentine — On November 22, 2022, FDA approved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
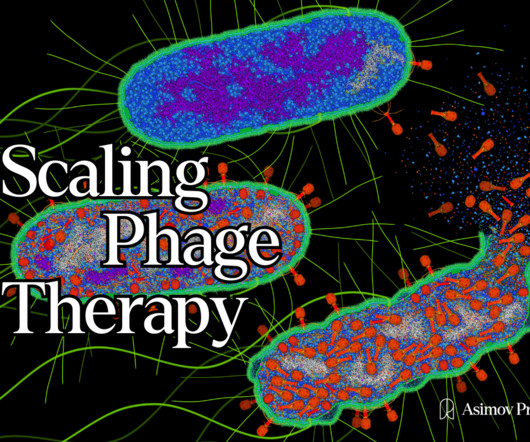
Tom Ireland writes about the companies and technologies that are reimagining phage therapy. Soon after its publication, scientists, journalists, and investors were revisiting ‘phage therapy’ as a promising alternative to our failing antibiotics. Read it on our website here. Illustration by David S. Fast forward to 2023.
Who better than people living with a condition to inform drug companies, physicians, academics, and the FDA on what it is like to live with their condition, what symptoms most impact their lives, what goes into their decision about whether to participate in a clinical trial, and what kind of treatment effects would be most meaningful to them?
The Countering Emerging Threats - Rapid Acquisition and Investigation of Drugs for Repurposing (CET RAIDR) program within the JPM Medical is designed to rapidly tackle known, unknown, and emerging threats by utilizing late-stage or licensed therapeutics. Repurposing is one such method. CBRN MCMs).
.– December 10, 2019 — Catalent, the leading global provider of advanced delivery technologies, development, and manufacturing solutions for drugs, biologics, gene therapies, and consumer health products, today announced that it has completed clinical production of Bridge Therapeutics Inc.’s
Food and Drug Administration (FDA) announced its acceptance of the Biologics License Application (BLA) for exa-cel. In recognition of the unmet need and medical urgency for innovative therapies in the sickle cell space, the FDA granted exa-cel Priority Review, with a formal decision expected by December 8, 2023.
Nasdaq: GMDA), an advanced cell therapy company committed to cures for blood cancers and serious hematologic diseases, today announced that the company conducted a Type B Meeting for omidubicel with the U.S. Food and Drug Administration (FDA) on Friday, December 11, 2020. BOSTON–( BUSINESS WIRE )– Gamida Cell Ltd.
NYSE: PFE) and BioNTech SE (Nasdaq: BNTX) today announced they have submitted a supplemental Biologics License Application (sBLA) to the U.S. Food and Drug Administration (FDA) to expand the approval of COMIRNATY® (COVID-19 Vaccine, mRNA) to include individuals ages 12 through 15 years. Pfizer Inc.
(Nasdaq: PASG), a genetic medicines company focused on developing transformative therapies for rare monogenic central nervous system (CNS) disorders, today announced that U.S. GM1 is a rare and often life-threatening CNS disorder with no approved disease-modifying therapies available. to 1 in 100,000 live births. “I About PBGM01.
The FDA's Biologics License Application (BLA) approvals in 2023 have marked a significant chapter in medical innovation, embodying precision and transformative therapies. A closer examination of the approvals, their numbers, and the trends they reveal unveils a dynamic landscape in biologics.
Food and Drug Administration (FDA) ’s focus appears to be on Emergency Use Authorizations (EUAs) for the Pfizer-BioNTech and Moderna COVID-19 vaccines, as the year wraps up there are still some PDUFA dates on the agency’s calendar. That collaboration was to develop and commercialize four oncology antibody biosimilar therapies.
(NASDAQ: REGN) today announced that the antibody cocktail casirivimab and imdevimab administered together (formerly known as REGN-COV2 or REGEN-COV2), a therapy currently being investigated for use in COVID-19 , has received Emergency Use Authorization (EUA) from the U.S. Food and Drug Administration (FDA). Schleifer, M.D.,
Food and Drug Administration (FDA) is plenty busy with COVID-19 vaccine Emergency Use Authorizations (EUAs) this month, but they’re also wrapping up the year with a few PDUFA dates for other therapies. Athenex and Almirall have a license deal with Almirall having the license to research, develop and commercialize the drug in the U.S.
. – Preclinical Data Underscore Treatment Potential for PBFT02 in Frontotemporal Dementia with Granulin (GRN) Mutations, a Devastating, Progressive Disorder Impacting Adults with No Approved Disease-Modifying Therapy Options. FTD is a debilitating form of early onset dementia that currently has no approved disease-modifying therapies. “We
Brevig, Senior Regulatory Device and Biologics Expert — On December 7, 2022, FDA’s Center for Biologics Evaluation and Research (CBER) and the Office of Tissues and Advanced Therapies (OTAT) held a town hall to answer questions related to cell therapy and tissue-engineered products chemistry, manufacturing, and controls (CMC).
Food and Drug Administration (FDA) are starting to pick up. The study enrolled 5,050 patients who received either vericiguat or placebo in combination with available heart failure therapies. The FDA granted the NDA Priority Review, which expedited the review from the typical 10 months to six months. Sarah Silbiger/Getty Images.
FDA advisory committees recommended just 50 percent of the 18 new therapies and indications they reviewed in 2020, the lowest rate since 2007, and the agency seems to be reserving the panels for more problematic applications, according to Prevision Policy, a Washington, D.C.-based based research firm.
Food and Drug Administration (FDA) has granted Fast Track designation to VTX-801, Vivet’s clinical-stage gene therapy for the treatment of Wilson Disease – a rare, genetic disorder that reduces the ability of the liver and other tissues to regulate copper levels, causing severe hepatic damage, neurological symptoms, and potentially death.
Food and Drug Administration (FDA) has a busy end of November planned, with numerous PDUFA dates to address. The FDA approved it under the brand name Gavreto on September 4. The drug is a once-daily oral precision therapy designed for highly potent and selective targeting of oncogenic RET alterations.
The FDA granted cilta-cel Breakthrough Therapy and Orphan Drug designations in 2019, and the therapy received a PRIME Priority Medicines designation from the European Commission (EC) that same year and an Orphan Drug designation from the EC in February 2020.
FDA Approves Danyelza (naxitamab-gqgk) for the Treatment of Neuroblastoma. Food and Drug Administration (“FDA”) has approved Danyelza (naxitamab-gqgk) 40mg/10ml. The product has received Priority Review, Orphan Drug, Breakthrough Therapy, and Rare Pediatric Disease designations from the FDA. NEW YORK, Nov.
Food and Drug Administration (FDA) in terms of PDUFA dates. Here’s a look at some of the upcoming target action dates on the FDA’s calendar. . The resubmission came after a meeting with the FDA in February 2020 and addresses issues raised by the agency’s Complete Response Letter (CRL) in November 2019. “We
The FDA issued straight-to-final guidance that provides sponsors of monoclonal antibody and other therapeutic protein COVID-19 treatments with recommendations for potency assays to ensure consistent product quality. The guidance does not cover vaccines, hyperimmune globulins, gene therapies, cell therapies or convalescent plasma.
Allecra, subject to the satisfaction of terms and conditions as set forth in the Exclusive Licensing Agreement, is to receive an upfront cash payment and is eligible to receive additional development and commercial milestone payments with an overall deal value of $78 million, in addition to royalties.
Lilly is offering donations of baricitinib to the Indian government through Direct Relief while simultaneously working with local Indian pharmaceutical companies to execute royalty-free voluntary licensing agreements to accelerate the manufacturing and distribution of the medicine in India during the pandemic.
Food and Drug Administration (FDA) Joint Arthritis Advisory Committee and Drug Safety and Risk Management Advisory Committee on tanezumab. We will continue to work with the FDA as the agency continues its review of our application.”. “We We will continue to work with the FDA as the agency continues its review of our application.”.
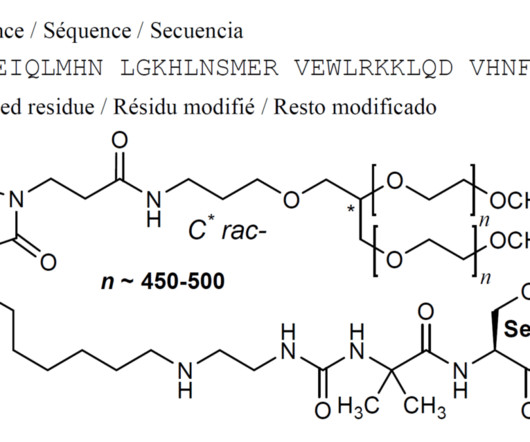
2222514-07-8 ] Palopegteriparatide , sold under the brand name Yorvipath , is a hormone replacement therapy used for the treatment of hypoparathyroidism. [1] 5] Study drug and conventional therapy were subsequently adjusted according to the albumin-corrected serum calcium levels. [5] 48,000) consisting of 34 amino acid residues.
Food and Drug Administration (FDA). Y-mAbs Therapeutics has a target action date of November 30 for its Biologics License Application (BLA) for Danyelza (naxitamab) for patients with relapsed/refractory high-risk neuroblastoma. The FDA approved the drug on November 24. Here’s a look.
Mr. Barrow has over a decade of experience leading drug development programs aimed at identifying and testing novel treatments in a wide range of disease conditions under FDA and EMA.
.
NEW YORK , Jan.
MindMed Co-CEO J.R.
About MindMed.
FDA accepts Dupixent ® (dupilumab) for priority review in adult s with prurigo nodularis. The target action date for the FDA decision is September 30, 2022. The FDA grants priority review to therapies that have the potential to provide significant improvements in the treatment, diagnosis or prevention of serious conditions.
Food and Drug Administration (FDA) has accepted for review the Prior Approval Supplement (PAS) to the Biologics License Application (BLA) for ABRILADA™ (adalimumab-afzb) as an interchangeable biosimilar to Humira® (adalimumab). The Biosimilar User Fee Act (BsUFA) goal date for an FDA decision is in Q4 2022. “An
a biotechnology company developing cell therapies that enable organ regeneration, announced today that the U.S Food and Drug Administration (FDA) has cleared its Investigational New Drug (IND) application. LyGenesis’s lead allogeneic cell therapy program is focused on liver regeneration for patients with end stage liver disease.
Food and Drug Administration (FDA) has accepted for review the supplemental Biologics License Application (sBLA) for Dupixent ® (dupilumab) as an add-on treatment for children aged 6 to 11 years with uncontrolled moderate-to-severe asthma. In the U.S., Dupixent is approved in the U.S.
Food and Drug Administration (FDA) granted Breakthrough Therapy designation for donanemab, Eli Lilly and Company’s (NYSE: LLY) investigational antibody therapy for Alzheimer’s disease (AD).
NASDAQ: AUPH / TSX:AUP) (“Aurinia” or the “Company”) today announced it has entered into a collaboration and license agreement with Otsuka Pharmaceutical Co., Food and Drug Administration (FDA) with an assigned Prescription Drug User Fee Act (PDUFA) target action date of January 22, 2021. VICTORIA, British Columbia & ROCKVILLE, Md.–(
To effectively navigate this ecosystem and expedite the development of new therapies, collaboration between the pharmaceutical industry and academia is proving increasingly vital. An example is the collaboration between Novartis and the University of Oxford to develop a gene therapy for spinal muscular atrophy, a rare genetic disease.
25, 2020 /PRNewswire/ — RhoVac, a clinical stage company today announced that the American FDA has granted Fast Track Designation to the company’s drug candidate, RV001.
More frequent written communication from FDA about such things as the design of the proposed clinical trials and use of biomarkers.
Quercis’ lead drug candidate acts as an antithrombotic with significantly lower risk of adverse events than existing therapies. Food and Drug Administration (FDA) approval for the prevention of VTE in all cancer types based on these two Phase 3 studies. ZUG, Switzerland , Jan. Chief Medical Officer of Quercis Pharma.
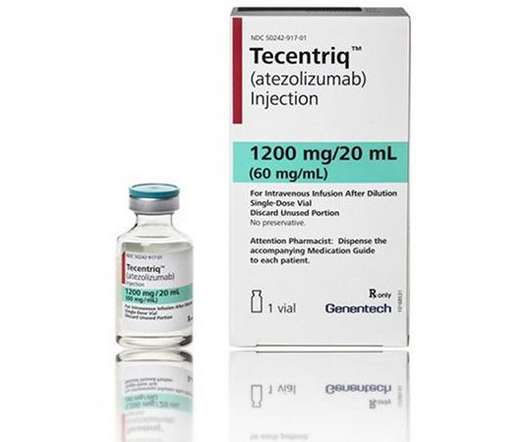
today announced that the US Food and Drug Administration (FDA) has accepted the company’s supplemental Biologics License Application (sBLA) and granted Priority Review for Tecentriq® (atezolizumab) as adjuvant treatment following surgery and platinum-based chemotherapy for people with non-small cell lung cancer (NSCLC) whose tumours express PD-L1?1%,
FDA ACCEPTS FOR PRIORITY REVIEW PFIZER’S APPLICATION FOR TICOVAC (TICK-BORNE ENCEPHALITIS VACCINE). In line with Priority Review designation, the FDA will target an action within six months of the application submission date, 3 with the anticipated Prescription Drug User Fee Act (PDUFA) action date expected for August 2021.
INDIANAPOLIS , May 4, 2021 /PRNewswire/ — Eli Lilly and Company (NYSE: LLY) is donating COVID-19 therapies to Direct Relief, enabling the humanitarian organization to provide COVID-19 therapies at no cost to low- and lower-middle-income countries most heavily impacted by the pandemic.
The Janssen Pharmaceutical Companies of Johnson & Johnson today announced the submission of a supplemental Biologics License Application (sBLA) to the U.S. Food and Drug Administration (FDA) seeking expanded approval of STELARA® (ustekinumab) to treat pediatric patients ages 5 years and older with juvenile psoriatic arthritis (jPsA).
Food and Drug Administration (FDA) did not approve the cancer drug liso-cel (lisocabtagene maraleucel) by the required Dec. The FDA accepted the Biologics License Application for ide-cel, an investigational B-cell maturation antigen (BCMA)-directed chimeric antigen receptor (CAR) T cell immunotherapy, under priority review in September.
The target action date for the FDA decision on this investigational use is August 3, 2022. FDA granted Breakthrough Therapy designation to Dupixent for the treatment of patients aged 12 years and older with EoE. If approved, Dupixent would be the first medicine available in the U.S. In September 2020, the U.S.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.














































Let's personalize your content