This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Who better than people living with a condition to inform drug companies, physicians, academics, and the FDA on what it is like to live with their condition, what symptoms most impact their lives, what goes into their decision about whether to participate in a clinical trial, and what kind of treatment effects would be most meaningful to them?
Authors: Matt Cooper, PhD, Executive Director, Therapeutic Strategy Lead, Oncology; Megan Morrison, Vice President, Asia Pacific Strategy Lead Adaptive trial designs have become essential in oncology, offering a flexible and efficient approach for conducting clinical trials.
Valentine — On November 22, 2022, FDA approved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
The FDA defines a serious condition as “a disease or condition associated with morbidity that has a substantial impact on day-to-day functioning.” Concerns have arisen over delays—sometimes spanning over 7–8 years—that may expose patients to risks before confirmatory trials are completed.
(NYSE: PFE) and BioNTech SE (Nasdaq: BNTX) today announced they have submitted a supplemental Biologics License Application (sBLA) to the U.S. Food and Drug Administration (FDA) to expand the approval of COMIRNATY® (COVID-19 Vaccine, mRNA) to include individuals ages 12 through 15 years.
Food and Drug Administration (FDA) announced its acceptance of the Biologics License Application (BLA) for exa-cel. In recognition of the unmet need and medical urgency for innovative therapies in the sickle cell space, the FDA granted exa-cel Priority Review, with a formal decision expected by December 8, 2023.
Food and Drug Administration (FDA) on Friday, December 11, 2020. During the meeting, the FDA provided encouraging feedback regarding the Phase 3 study of omidubicel pertaining to the pre-specified primary and secondary endpoints. For more information on clinical trials of omidubicel, please visit www.clinicaltrials.gov.
FDA this year. “Rolontis,” a treatment for neutropenia that had its technology licensed out to Spectrum Pharmaceuticals, Inc. and “Oraxol,” which was licensed out to Athenex, Inc. “Rolontis,” a treatment for neutropenia that had its technology licensed out to Spectrum Pharmaceuticals, Inc.
While traditionally conducted during Phase 2 or later, the FDA has recently been requesting data sooner in the development process, making it critical to implement proactive AME studies to lay the groundwork for advanced phases of your clinical program. To accomplish this, we pay careful attention to both dosing and sample collection.
Food and Drug Administration (FDA) ’s focus appears to be on Emergency Use Authorizations (EUAs) for the Pfizer-BioNTech and Moderna COVID-19 vaccines, as the year wraps up there are still some PDUFA dates on the agency’s calendar. Although much of the U.S. Read on for this week’s. MacroGenics’ Margetuximab for Metastatic HER2+ Breast Cancer.
Mr. Barrow has over a decade of experience leading drug development programs aimed at identifying and testing novel treatments in a wide range of disease conditions under FDA and EMA.
.
NEW YORK , Jan.
MindMed Co-CEO J.R.
Food and Drug Administration (FDA) has designated as a Fast Track development program the investigation of Brilacidin as a potential treatment for COVID-19. Positive results were also observed in a Phase 2 Proof-of-Concept trial treating patients locally with Brilacidin for Ulcerative Proctitis/Ulcerative Proctosigmoiditis (UP/UPS).
Food and Drug Administration (FDA) has a busy end of November planned, with numerous PDUFA dates to address. The FDA approved it under the brand name Gavreto on September 4. Patent Trial and Appeal Board (PTAB) had instituted inter partes review (IPR) against U.S. The data was based on the Phase III KEYNOTE-355 trial.
Food and Drug Administration (FDA) is plenty busy with COVID-19 vaccine Emergency Use Authorizations (EUAs) this month, but they’re also wrapping up the year with a few PDUFA dates for other therapies. Vibegron could be a potentially important and differentiated new oral treatment, if approved by the FDA, for patients suffering with OAB.”.
Food and Drug Administration (FDA) are starting to pick up. The NDA was built on data from the Phase III VICTORIA trial. We will continue to collaborate with the FDA during their review process and in parallel build our commercial readiness for a potential approval and commercial launch in the first quarter of 2021.”.
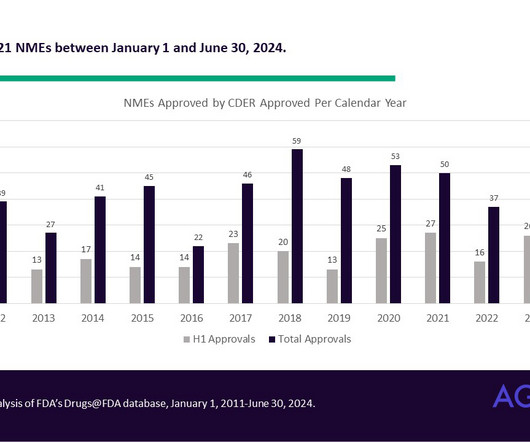
Halfway there: Novel drug approvals and their supportive clinical trials so far in 2024 In the first half of 2024, the FDA’s Center for Drug Evaluation and Research (CDER) approved 21 novel drug products. CDER is the FDA office in charge of reviewing pharmaceuticals and therapeutic biologics.
Food and Drug Administration (FDA) has cleared an investigational new drug (IND) application for the company’s lead product candidate, PBGM01, an adeno-associated virus (AAV)-delivery gene therapy that is being studied for the treatment of infantile GM1 gangliosidosis (GM1). president and chief executive officer of Passage Bio.
Food and Drug Administration (FDA). Y-mAbs Therapeutics has a target action date of November 30 for its Biologics License Application (BLA) for Danyelza (naxitamab) for patients with relapsed/refractory high-risk neuroblastoma. The submission was based on the safety and efficacy data of the pivotal Phase II trials 201 and 12-230.
Many of our team at the FDA are parents and grandparents themselves, and our team shares an equivalent concerns as many in our country about protecting our loved ones from COVID-19. The FDA will work closely with each manufacturer to make sure this data analysis is strong and meets regulatory standards.
Food and Drug Administration (FDA) in terms of PDUFA dates. Here’s a look at some of the upcoming target action dates on the FDA’s calendar. . The company is also still conducting a Phase III trial in the adult ADHD patient population. The sNDA was built on data from the Phase III CANDOR trial.
The Company will initiate its trial during the first quarter of 2021 to investigate the efficacy of Berubicin in adults with GBM who have failed first-line therapy. and 2 trials planned by our sublicensee WPD in Poland. . and 2 trials planned by our sublicensee WPD in Poland.
About CNS Pharmaceuticals, Inc.
FDA Approves Danyelza (naxitamab-gqgk) for the Treatment of Neuroblastoma. Food and Drug Administration (“FDA”) has approved Danyelza (naxitamab-gqgk) 40mg/10ml. Continued approval for this indication may be contingent upon verification and description of clinical benefits in a confirmatory trial. NEW YORK, Nov.
Maintaining complete and compliant documentation while managing the complex processes and interactions of clinical trial conduct is complicated. Investigator Site Files The ISF contains essential documents permitting evaluation of a clinical trial’s conduct. Form FDA 1572 The Form FDA 1572 is unique to U.S.-based
A regulatory binder is essential for managing clinical trial documents, ensuring regulatory compliance, and facilitating audits. It organizes critical documents; provides easy access for trial monitors, auditors, and regulatory authorities; and serves as a reference for the research team.
Allecra, subject to the satisfaction of terms and conditions as set forth in the Exclusive Licensing Agreement, is to receive an upfront cash payment and is eligible to receive additional development and commercial milestone payments with an overall deal value of $78 million, in addition to royalties.
Food and Drug Administration (FDA) has granted Fast Track designation to VTX-801, Vivet’s clinical-stage gene therapy for the treatment of Wilson Disease – a rare, genetic disorder that reduces the ability of the liver and other tissues to regulate copper levels, causing severe hepatic damage, neurological symptoms, and potentially death.
FDA accepts Dupixent ® (dupilumab) for priority review in adult s with prurigo nodularis. The target action date for the FDA decision is September 30, 2022. The safety results from these trials were generally consistent with the known safety profile of Dupixent in atopic dermatitis.
NIH trial seeks answers. (
Third time’s the charm as Heron wins FDA nod for non-opioid anesthetic Zynrelef ( Endpoints ).
Amgen, AstraZeneca bolster their case for breakthrough asthma program as FDA considers taking up a review ( Endpoints ).
In Focus: International.
Medtech.
FDA fiscal year in review: New drug approvals in the wake of the pandemic and legislative reforms AgencyIQ analyzed CDER’s novel drug approvals in Fiscal Year 2023, identifying a recovery in approval numbers as the agency resumes a new normal following the pandemic. The FDA also refers to novel products as “New Molecular Entities,” or NMEs.
More than a month has passed since AstraZeneca’s phase 3 COVID-19 vaccine trial was paused due to a potential adverse event, but few solid details have emerged from ongoing safety reviews by the FDA and the UK drugmaker. Neither the company nor the FDA responded to repeated requests for comment on this report. James Miessler.
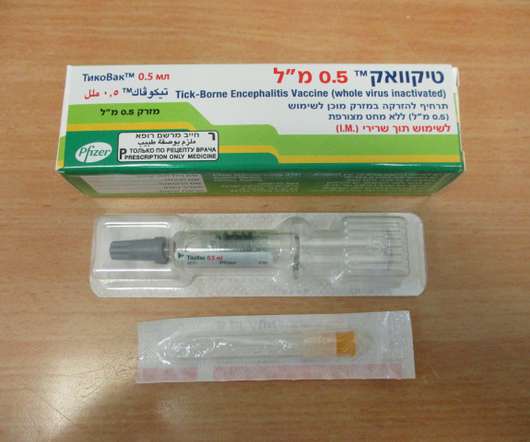
Food and Drug Administration (FDA) has approved TICOVAC (tick-borne encephalitis (TBE) vaccine) for active immunization to prevent TBE in individuals 1 year of age and older. 1 TICOVAC is the only FDA-approved vaccine to help protect U.S. Following today’s FDA approval, the U.S. in 1-15 year olds and 98.7-100%
5] History The effectiveness of palopegteriparatide was evaluated in a 26-week, randomized, double-blind, placebo-controlled trial that enrolled 82 adults with hypoparathyroidism. [5] 5] The FDA granted the application for palopegteriparatide orphan drug and priority review designations. [5] 4] It is a parathyroid hormone analog. [1]
Patient dosing has begun in a Phase III clinical programme investigating GlaxoSmithKline’s 5-in-1 meningitis (MenABCWY) vaccine candidate compared to licensed meningococcal vaccines, Bexsero and Menveo. Invasive Meningococcal Disease (IMD) is uncommon, with country-specific reported cases ranging from 0.1
The FDA has also granted precedence review to the company’s sBLA for Kymriah in adult cases with r/ r FL. The nonsupervisory cessions are grounded on positive data from the vital Phase II ELARA trial, which delved the efficacity and safety of Kymriah in adult cases with r/ r FL.
The Janssen Pharmaceutical Companies of Johnson & Johnson today announced the submission of a supplemental Biologics License Application (sBLA) to the U.S. Food and Drug Administration (FDA) seeking expanded approval of STELARA® (ustekinumab) to treat pediatric patients ages 5 years and older with juvenile psoriatic arthritis (jPsA).
FDA ACCEPTS FOR PRIORITY REVIEW PFIZER’S APPLICATION FOR TICOVAC (TICK-BORNE ENCEPHALITIS VACCINE). In line with Priority Review designation, the FDA will target an action within six months of the application submission date, 3 with the anticipated Prescription Drug User Fee Act (PDUFA) action date expected for August 2021.
Food and Drug Administration (FDA) has broadened the Emergency Use Authorization (EUA) for baricitinib to allow for treatment with or without remdesivir, whereas the EUA was previously restricted to use only in combination with remdesivir. . Baricitinib now authorized for emergency use as monotherapy. Wesley Ely, M.D., In the U.S.,
All clinical trial or marketing applications submitted to the FDA must include a form that summarizes the content of the submission and any relevant information on the sponsor and drug for the reviewers. Periodically, the FDA updates these forms to enhance their usability and incorporate relevant content to help sponsors and reviewers.
We remain committed to further progressing our trial preparations, as we look forward to initiating a U.S. Phase 2 trial for Berubicin during the first quarter of 2021.”
In addition to its manufacturing efforts, the Company has also made progress in its clinical trial preparations.
Food and Drug Administration (FDA) has accepted for review the supplemental Biologics License Application (sBLA) for Dupixent ® (dupilumab) as an add-on treatment for children aged 6 to 11 years with uncontrolled moderate-to-severe asthma. Detailed results from this Phase 3 trial will be published later this year. In the U.S.,
This has been borne out in a Phase 2 clinical trial, which demonstrated evidence that prophylactic administration of Quercis’ investigational drug reduced levels of key markers of coagulation, without observed VTE or major bleeding events 2. ZUG, Switzerland , Jan. Chief Medical Officer of Quercis Pharma.
The target action date for the FDA decision on this investigational use is August 3, 2022. The sBLA is supported by data from two Phase 3 trials evaluating the efficacy and safety of Dupixent 300 mg weekly in patients aged 12 years and older with EoE ( Part A and Part B ), and data from an active long-term extension trial.
1 Ethical concerns surrounding the use of animal studies is increasing, especially considering 90 percent of drug candidates fail in clinical trials. 2 Therefore, the scientific community is researching and developing efficient ways to test compounds without the use of animals, to avoid unsuccessful outcomes in clinical trials.
United Kingdom Medicines and Healthcare Products Regulatory Agency authorized Clinical Trial Application.
With these important regulatory clearances for our first-in-human clinical trial for INZ-701 in subjects with ENPP1 deficiency, we have transitioned from a research-stage to a clinical-stage company.
BOSTON, Jan.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.













































Let's personalize your content