This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
The study examines the frequency, distribution, prevalence, and diversity of oxygen atoms in a dataset of 2049 small molecules approved by the FDA and other agencies. The analysis focuses on various types of oxygen atoms, including sp 3 , sp 2 -hybridized, ring, and nonring.
Lenz, Principal Medical Device Regulation Expert — For several years, FDA has requested that sponsors of drug or biologic led combination products identify essential performance requirements (EPRs) related to the device constituent in their applications. By Adrienne R. does not use this term. does not use this term.
Together, the tools estimate how a drug may impact diverse outcomes of interest to drug developers: general cellular health, pharmacokinetics, and heart and liver function. Since the FDA released these datasets, we wondered if we could use them to predict toxicity using machine learning," said Seal.
The Importance of Scientific Expertise Generic drug development involves a deep understanding of the scientific principles underlying drug development, including pharmacology, pharmacokinetics, and bioequivalence. Scientific experts in these fields are critical in ensuring that generic drugs are developed to meet the required standards.
Lenacapavir (LEN), a long-acting injectable, is the first approved human immunodeficiency virus type 1 capsid inhibitor and one of a few FDA-approved drugs that exhibit atropisomerism.
These protocols were then used to determine the plasma pharmacokinetics of agmatine and the extent of distribution to the CNS. Precision and accuracy of the protocol met Food and Drug Administration (FDA) standards in surrogate matrix, as well as in corrected concentrations in appropriate matrices.
On October 31, 2024, FDA issued its final version of the ICH M13A guidance for industry, titled M13A Bioequivalence for Immediate-Release Solid Oral Dosage Forms.
Secondary VAS and pharmacokinetic (PK) endpoints and adverse events were assessed. Drug Liking and all other VAS outcomes were greatest for nabilone 3mg and 6mg, which is a currently FDA-approved medication. Three doses of lenabasum (20, 60, and 120mg) were compared to placebo, and nabilone (3 and 6mg).
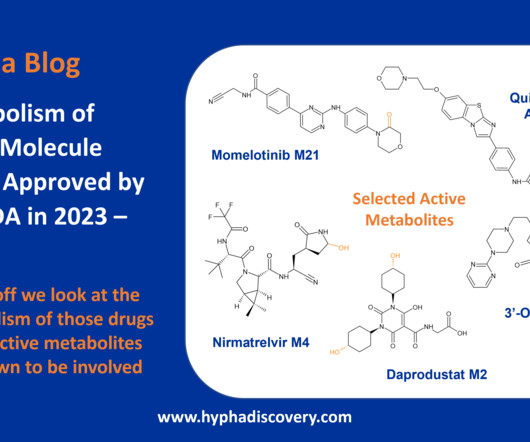
Table 1: Small molecule drugs approved by the FDA in 2023 with reported involvement of phase II mechanisms In vitro : In vivo differences Incubation of the SGLT2 (sodium-glucose co-transporter-2) inhibitor bexagliflozin in human liver microsomes points to metabolism through both oxidation and glucuronidation to 6 main metabolites.
It is critical that the nonclinical program outlined in the PIND briefing document is presented in a manner that allows FDA to provide relevant input on the required IND-enabling studies. For example, if the nonclinical data presented in the PIND briefing document lacks information on the potential toxicity profile of the drug (e.g.,
The FDA has approved two vaccines, and three vaccines are available for emergency use, to prevent COVID-19 and the serious clinical outcomes associated with COVID-19, including hospitalization and death. The FDA urges the public to get vaccinated and receive a booster when eligible. The FDA granted approval to Gilead Sciences Inc.
Food and Drug Administration (FDA) ’s focus appears to be on Emergency Use Authorizations (EUAs) for the Pfizer-BioNTech and Moderna COVID-19 vaccines, as the year wraps up there are still some PDUFA dates on the agency’s calendar. We look forward to working with the FDA to bring ABP 798 to market.”. Although much of the U.S.
The study demonstrated favorable proof-of-concept for LYT-100’s tolerability and pharmacokinetic (PK) profile, which will also enable twice-a-day (BID) dosing of LYT-100 in future studies. The therapeutic dose of pirfenidone approved by the US Food and Drug Administration (FDA) for the treatment of IPF is 801 mg three times a day.
A weekly update on new drug approvals and indications from the US Food and Drug Administration (FDA). . It also provided supporting pharmacokinetic data demonstrating the opioid antagonist’s safety and efficacy. . New approvals. Higher-dose Kloxxado nasal spray cleared for countering opioid overdose.
Metabolism of 2023 FDA Approved Small Molecules – PART 1 By Julia Shanu-Wilson 2023 was a fruitful year for drug approvals by the FDA, with a crop of 34 small molecules out of a total of 55 new drugs [1]. are major metabolites according the FDA Metabolites in Safety Testing guidelines). 2] Iversen et al.,
While the type, number, and design of these studies vary based on product-specific characteristics, IND-enabling packages submitted to the FDA generally include key information about the pharmacology, pharmacokinetics, and toxicology of the product. All these studies need to be performed under GLP.
Food and Drug Administration (FDA) has accepted for review the Prior Approval Supplement (PAS) to the Biologics License Application (BLA) for ABRILADA™ (adalimumab-afzb) as an interchangeable biosimilar to Humira® (adalimumab). The Biosimilar User Fee Act (BsUFA) goal date for an FDA decision is in Q4 2022. “An
It also displayed favourable pharmacokinetics (PK) and is well tolerated in non-human primates (NHP) at exposure levels above those projected to be efficacious. ZW191 also displayed favourable pharmacokinetics (PK) and is well tolerated in non-human primates (NHP) at exposure levels above those projected to be efficacious.
Food and Drug Administration (FDA) in terms of PDUFA dates. Here’s a look at some of the upcoming target action dates on the FDA’s calendar. . The resubmission came after a meeting with the FDA in February 2020 and addresses issues raised by the agency’s Complete Response Letter (CRL) in November 2019. “We
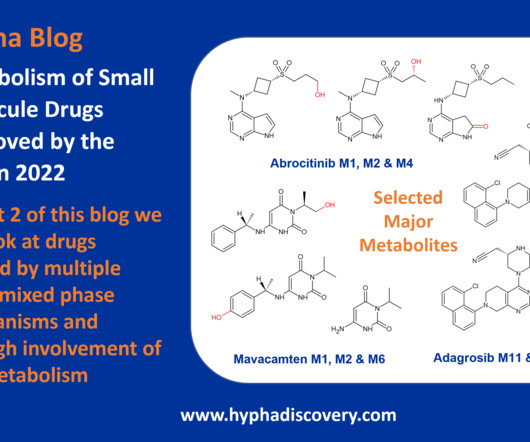
Metabolism of 2022 FDA approved small molecule drugs part 2 Mixing it Up By Julia Shanu-Wilson In Part 1 of this topic we looked at metabolism of the small molecule drugs approved by the FDA in 2022 that were mediated by CYP3A4. 16 This concludes our exploration of metabolism of last year’s FDA approved small molecule drugs.
His presentation covered various aspects of oncology clinical programs, focusing on study design trends, with reference to both the recently implemented FDA Project Optimus guidance and studies we have seen from sponsors.
In new contract notices, FDA seeks help on user fee commitments and moving opioid, oncology priorities forward This week, the FDA issued four new contract notices that highlight agency priorities in meeting user fee commitments, ongoing work in oncology dose optimization, and continued commitment to addressing the opioid crisis.
FDA looking to beef up its remote assessments with artificial intelligence According to a new FDA contract notice, the agency is interested in procuring a new artificial intelligence-based Optical Character Recognition system to extract data from PDFs obtained during remote regulatory assessments. What does the FDA want to do, anyways?
FDA updates guidance on developing drugs for Covid-19, replacing pandemic-era version Last week, FDA published the third update to its guidance on the development of products to prevent or treat Covid-19. The FDA also implemented new flexibilities for certain regulated products and processes, typically via enforcement discretion.
Since 1962, the FD&C Act has authorized FDA to require that sponsors of clinical trials submit data from “preclinical tests (including tests on animals)” in order to demonstrate that their drug is safe enough to advance to testing in humans. For more on FDORA’s other provisions, see HPM’s complete summary here ). 21 U.S.C. § 355(i)(1)(A).
“Population pharmacokinetic/pharmacodynamic modelling of eplontersen, an antisense oligonucleotide in development for transthyretin amyloidosis” British Journal of Clinical Pharmacology. S2CID 250989659. ^ “Eplontersen: FDA-Approved Drugs” U.S. Food and Drug Administration (FDA). Ionis Pharmaceuticals.
The US FDA Modernisation Act 2.0., In spite of current success and possibility to be a successful cell technology model, the integration of organ-on-chips into drug development process needs more optimisation to be validated for FDA approval. Prior to this European Union Parliament, in 2021, voted for animal testing phase out.
Submission supported by comprehensive analytical and clinical data from new Phase I bridging pharmacokinetics study Adalimumab’s high-concentration 100 mg/mL formulation aims to provide an enhanced yet familiar experience for patients Submission builds on Sandoz’ well established biosimilar immunology portfolio in Europe.
On July 31, 2024, the US Food and Drug Administration (FDA) announced Fiscal Year 2025 (FY2025) Prescription Drug User Fee Amendments of 2022 (PDUFA VII) fee rates for the review of human drug and biological product applications along with prescription drug program fees. FDA-2024-N-0007. FDA-2023-N-2850. 89 FR 61474.
A review of FDA’s Postmarketing Requirements and Commitments database reveals that one of the most common reasons FDA requires postmarketing studies is to assess the impact of a drug on maternal and fetal outcomes when taken by pregnant women. Additionally, this review excluded pharmacokinetic and animal studies.
Nasdaq GILD) moment blazoned new data from an interim analysis of its ongoing, Phase2/3 single arm, open- marker study to estimate the safety, tolerability and pharmacokinetics of Veklury ® (remdesivir) in pediatric cases rehabilitated with COVID-19 with periods ranging from 28 days to lower than 18 times. Gilead Lores,Inc.
FDA offers a status check on its diversity in research provisions, one year post-FDORA At a workshop on FDA’s implementation of new statutory requirements for diversity in clinical research, agency and industry representatives gave a status update on implementation.
The FDA EUA submission is based on an interim analysis of efficacy and safety data from the Phase 3 COMET-ICE (COVID-19 Monoclonal antibody Efficacy Trial – Intent to Care Early) trial, which evaluated VIR-7831 as monotherapy for the early treatment of COVID-19 in adults at high risk of hospitalisation.
In this blog, we explain the role of clinical pharmacology in drug development and demonstrate how the right strategy can accelerate development under the US Food and Drug Administration (FDA) 505(b)(1) and 505(b)(2) New Drug Application (NDA) pathways. Author: Jayesh Patel , Principal Scientist, Pharmacokinetics
The team must present data to the IDSMB accurately and promptly, especially when multiple trial arms progress at different rates, demanding comprehensive data management, including safety, pharmacokinetic (PK), or pharmacodynamic (PD) data, and statistical inputs. These include: FDA (U.S.)
The application was submitted under the FDA’s accelerated approval program. The review is being conducted under Project Orbis, an initiative of the FDA Oncology Center of Excellence (OCE), which provides a framework for concurrent submission and review of oncology products among international partners. In 2019, the U.S.
The trial, which will explore the pharmacokinetics and safety of ATX01 in healthy volunteers, is due to start in January 2021. In parallel, AlgoTx firmed-up ATX01’s development pathway via a pre-IND consultation with the FDA and obtained an Orphan Drug Designation from the FDA to explore ATX01’s activity in erythromelalgia.
Food and Drug Administration (FDA) has approved long-acting atypical antipsychotic INVEGA HAFYERA™ (6-month paliperidone palmitate), the first-and-only twice-yearly injectable for the treatment of schizophrenia in adults. You may report side effects to FDA at 1-800-FDA-1088. shuffling walk.
Parallels between pharmacokinetic (PK) analyses for CGTs and immunotherapies, conducted through PCR-based assays and plate-bound antibody assays, respectively, can inform a successful approach. Food and Drug Administration (FDA). However, key differences between PCR assays and immunotherapeutic assays must be considered.
AI has the capability to analyze and forecast the pharmacokinetic (PK) profiles of drugs following their administration. Additionally, AI can examine the correlation between drug exposure and response, including the effect of variables, thus ensuring optimal dose selection and dosing regimens for clinical research.
BY AMANDA CONTI, ALEXANDER GAFFNEY, MS, RAC SEP 18, 2023 9:24 PM CDT Background: FDA’s standards for evidence When seeking approval from the FDA, companies are required to demonstrate that their product is safe and effective when used as intended. In one landmark case, Warner-Lambert Co.
Food and Drug Administration (FDA) approved DALVANCE® (dalbavancin) for the treatment of acute bacterial skin and skin structure infections (ABSSSI) in pediatric patients from birth. AbbVie (NYSE: ABBV) today announced that the U.S.
We appreciated the opportunity to share our data with the Advisory Committee, and we will continue to work with the FDA as it completes its review of our application.”. FDA Advisory Committees provide non-binding recommendations for consideration by the FDA. CAMBRIDGE, Mass. and TOKYO, Nov. About Alzheimer’s Disease.
Molecular Weight: 631.700 FDA APPROVED, To treat moderately to severely active ulcerative colitis in adults, 10/12/2023 Velsipity Etrasimod , sold under the brand name Velsipity , is a medication that is used for the treatment of ulcerative colitis (UC). [1] Food and Drug Administration (FDA). 1] It is taken by mouth. [1]
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.









































Let's personalize your content