This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
These protocols were then used to determine the plasma pharmacokinetics of agmatine and the extent of distribution to the CNS. Precision and accuracy of the protocol met Food and Drug Administration (FDA) standards in surrogate matrix, as well as in corrected concentrations in appropriate matrices.
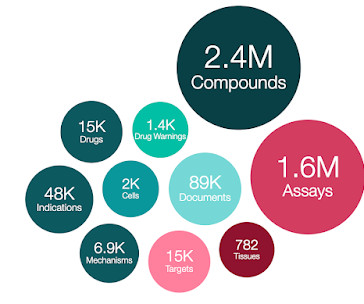
71 out of the 882 newly added EMA drugs are only authorised by EMA, rather than from other regulatory bodies e.g. FDA. Progress has been made towards standardising drug and clinical candidate pref_names (in MOLECULE_DICTIONARY.PREF_NAME) whereby an approved drug name (FDA /EMA) is assigned in the first instance, if available.
A weekly update on new drug approvals and indications from the US Food and Drug Administration (FDA). . It also provided supporting pharmacokinetic data demonstrating the opioid antagonist’s safety and efficacy. . Farxiga received fast track, breakthrough therapy and priority review designations for the current indication. .
Food and Drug Administration (FDA) ’s focus appears to be on Emergency Use Authorizations (EUAs) for the Pfizer-BioNTech and Moderna COVID-19 vaccines, as the year wraps up there are still some PDUFA dates on the agency’s calendar. That collaboration was to develop and commercialize four oncology antibody biosimilar therapies.
It also displayed favourable pharmacokinetics (PK) and is well tolerated in non-human primates (NHP) at exposure levels above those projected to be efficacious. ZW191 also displayed favourable pharmacokinetics (PK) and is well tolerated in non-human primates (NHP) at exposure levels above those projected to be efficacious.
Food and Drug Administration (FDA) in terms of PDUFA dates. Here’s a look at some of the upcoming target action dates on the FDA’s calendar. . The resubmission came after a meeting with the FDA in February 2020 and addresses issues raised by the agency’s Complete Response Letter (CRL) in November 2019. “We
The study demonstrated favorable proof-of-concept for LYT-100’s tolerability and pharmacokinetic (PK) profile, which will also enable twice-a-day (BID) dosing of LYT-100 in future studies. The therapeutic dose of pirfenidone approved by the US Food and Drug Administration (FDA) for the treatment of IPF is 801 mg three times a day.
His presentation covered various aspects of oncology clinical programs, focusing on study design trends, with reference to both the recently implemented FDA Project Optimus guidance and studies we have seen from sponsors.
Food and Drug Administration (FDA) has accepted for review the Prior Approval Supplement (PAS) to the Biologics License Application (BLA) for ABRILADA™ (adalimumab-afzb) as an interchangeable biosimilar to Humira® (adalimumab). The Biosimilar User Fee Act (BsUFA) goal date for an FDA decision is in Q4 2022. “An
New Indication for Amgen’s Fifth FDA-approved Biosimilar. The randomized, double-blind, comparative clinical study compared the efficacy, safety, pharmacokinetics and immunogenicity of RIABNI versus rituximab reference product (RP) in patients with moderate to severe RA. Now Approved to Treat All Available Rituxan ® Indications.
7 These inhibitors have faced challenges such as dose-limiting toxicity and poor pharmacokinetics, but geldanamycin ADCs have demonstrated increased survival in mice. Dual payload ADCs As effective as therapies have been in treating solid and haematological cancers, tumour heterogeneity and resistance remain major clinical challenges.
Mobocertinib is the first oral therapy specifically designed to selectively target EGFR Exon20 insertion mutations. The application was submitted under the FDA’s accelerated approval program. and around the globe.”. Takeda has established an Expanded Access Program (EAP) ( NCT04535557 ) for patients in the U.S. In 2019, the U.S.
Cell and gene therapies (CGTs) are one of the fastest growing areas in human therapeutics. Since chimeric antigen receptor T cell (CAR-T) therapy was first approved in 2017, there has been a marked increase of cell and gene therapy studies resulting in significant changes in the way diseases are treated as well as patient outcomes.
“Characteristics of Patients with Hereditary Transthyretin Amyloidosis-Polyneuropathy (ATTRv-PN) in NEURO-TTRansform, an Open-label Phase 3 Study of Eplontersen” Neurology and Therapy. S2CID 250989659. ^ “Eplontersen: FDA-Approved Drugs” U.S. Food and Drug Administration (FDA). Ionis Pharmaceuticals.
INDIANAPOLIS , May 4, 2021 /PRNewswire/ — Eli Lilly and Company (NYSE: LLY) is donating COVID-19 therapies to Direct Relief, enabling the humanitarian organization to provide COVID-19 therapies at no cost to low- and lower-middle-income countries most heavily impacted by the pandemic.
Sanofi and Swedish Orphan Biovitrum AB (publ) (Sobi ® ) (STO:SOBI) today announced positive topline results from the pivotal XTEND-1 Phase 3 study evaluating the safety, efficacy and pharmacokinetics of efanesoctocog alfa (BIVV001) in previously treated patients ? The US FDA granted Fast Track Designation in February 2021.
FDA’s newest draft guidance lays out considerations for developing treatments for diabetic foot infections In a new draft guidance document released within weeks of a related clinical practice guideline, the FDA provides considerations for developing therapies to treat diabetic foot infections, focusing on Phase 3 efficacy trials.
Psychedelics Psychedelic therapy (or psychedelic-assisted therapy) refers to the use of psychedelic drugs, such as psilocybin, MDMA, LSD, ketamine, and ayahuasca, to treat mental disorders, especially those that have no effective treatments available or are treatment resistant.
PAXLOVID™ (nirmatrelvir [PF-07321332] tablets and ritonavir tablets) is the first oral therapy specifically designed to combat COVID-19 to be evaluated in a pediatric clinical study PAXLOVID is currently authorized under U.S. The safety and effectiveness of PAXLOVID have not yet been directly established in pediatric patients.
In the absence of a clinical trial result or FDA label to point to, how does one create the case and target product profile (TPP) around a new target? with gene editing or gene therapy, enzyme replacement therapy), agonism (e.g., in liver, in CNS)? with antibodies), or correction (e.g., in the case of CFTR for Trikafta).
Food and Drug Administration (FDA) approved DALVANCE® (dalbavancin) for the treatment of acute bacterial skin and skin structure infections (ABSSSI) in pediatric patients from birth. 20% reduction in lesion size compared to baseline and no receipt of rescue antibacterial therapy for children 3 months and older.
Molecular Weight: 631.700 FDA APPROVED, To treat moderately to severely active ulcerative colitis in adults, 10/12/2023 Velsipity Etrasimod , sold under the brand name Velsipity , is a medication that is used for the treatment of ulcerative colitis (UC). [1] Food and Drug Administration (FDA). 1] It is taken by mouth. [1] April 2023).
of new cancer drugs tested in Phase I were likely to receive Food and Drug Administration (FDA) approval. cell and gene therapies), with other therapeutic areas then pushing it further. Platform-type studies aiming to open and close cohorts quickly based on surrogate endpoints can help explore these targeted therapies more efficiently.
BY AMANDA CONTI, ALEXANDER GAFFNEY, MS, RAC SEP 18, 2023 9:24 PM CDT Background: FDA’s standards for evidence When seeking approval from the FDA, companies are required to demonstrate that their product is safe and effective when used as intended. In one landmark case, Warner-Lambert Co.
Food and Drug Administration (FDA) clearance of an Investigational New Drug (IND) application for SLV213 for the treatment of COVID-19 and has dosed the first subjects in a Phase 1 clinical study. Preclinical data show SLV213 is a potent inhibitor of SARS-CoV-2 infection. SAN DIEGO–( BUSINESS WIRE )– Selva Therapeutics, Inc.
We appreciated the opportunity to share our data with the Advisory Committee, and we will continue to work with the FDA as it completes its review of our application.”. FDA Advisory Committees provide non-binding recommendations for consideration by the FDA. CAMBRIDGE, Mass. and TOKYO, Nov. About Alzheimer’s Disease.
Biosimilars help patients to gain broader access to effective and high-quality treatments that improve their disease therapies,” said Rebecca Guntern, Head of Region Europe, Sandoz. The study met all its primary objectives, demonstrating comparable pharmacokinetics and showing similar safety and immunogenicity between the two concentrations.
The Phase I trial was performed to evaluate safety, tolerability, and pharmacokinetics in a group of 80 healthy participants in a two-part, double-blind, placebo-controlled study. The results of the trial showed a well-characterized pharmacokinetic profile, demonstrating dose proportionality over the ranges studied.
Food and Drug Administration (FDA) has extended the Prescription Drug User Fee Act (PDUFA) date for review of the Company’s Biologics License Application (BLA) seeking accelerated approval of pegunigalsidase alfa (PRX–102) for the proposed treatment of adult patients with Fabry disease.
About Pegunigalsidase Alfa.
percent) in patients taking therapy and 36 events (7.0 Bamlanivimab and etesevimab together also demonstrated statistically significant improvements on all key secondary endpoints, providing strong evidence that the therapy reduced viral load and accelerated symptom resolution. It remains under review by the FDA.
“At Pfizer, we have a strong heritage in, and commitment to, fighting infectious diseases, most recently evidenced by our delivery of the first authorized vaccine and oral therapy to combat COVID-19,” said Albert Bourla, Chairman and Chief Executive Officer, Pfizer. Food and Drug Administration (FDA). Sapir, CEO, ReViral.
Food and Drug Administration (“FDA”) for the treatment of agitation associated with delirium. Treatment choices are limited, and commonly used off-label therapies are not always effective or may result in prolonged, deep sedation. NEW HAVEN, Conn., 26, 2020 (GLOBE NEWSWIRE) — BioXcel Therapeutics, Inc. (“BTI”
3] While MET inhibitors have recently received accelerated approval in this setting in some regions, the vast majority of patients eventually acquire resistance to these therapies, thus underscoring the need for new treatment options. [4] METex14 mutations are found in approximately three percent of patients with NSCLC. [2] 4] , [5] , [6].
Of the 22 patients enrolled in the BRIDGE study, the majority of treatment emergent adverse events were mild or moderate in severity, with two patients (9.1%) withdrawing from the therapy due to hypersensitivity reaction that was resolved. The most common moderate treatment emergent adverse events were nasopharyngitis, headache and dyspnea.
The FDA and oncology: 2023 year in review As we round the corner into the last few weeks of 2023, AgencyIQ has taken a look back at a very busy year for the FDA’s oncology staff, and for sponsors. Since its inception, OCE has initiated an extensive list of initiatives and has taken the lead on over forty guidances published by FDA.
FDA finalizes new guidance on nonclinical assessment of potential immunotoxicity Late last week the FDA released a final guidance document containing recommendations on the nonclinical assessment of potential immunotoxicity for drugs and certain biologics.
phase I trial is a randomized, double-blind, placebo-controlled, single and multiple dose escalation study to evaluate safety, tolerability, pharmacokinetics and pharmacodynamics (biomarkers – FGF19 and C4) of ASC42 in healthy subjects. Food and Drug Administration (FDA) for NASH. ASC42 is an in-house developed?novel
The clinical responses were sustained by maintenance therapy with belimumab, an antibody to B-cell activating factor. The secondary outcome involves pharmacokinetic endpoints. The FDA has set an action date of July 7, 2021. The cells secrete auto-antibodies, but do not respond to standard immunosuppression. Receptor Inhibitors.
This small molecule therapy is presently in Phase 1 clinical trial for mild to moderate Alzheimer’s disease (AD), which is supported by a NIA R01 grant in healthy aged volunteers. Alzheimer’s is a disease of aging, the greatest risk-factor, but there is not a single therapy available to halt or reverse this chronic neurodegenerative disorder.
Food and Drug Administration (FDA) had granted Breakthrough Therapy designation for donanemab based on the Phase 2 data. Additionally, pharmacokinetic/pharmacodynamic modeling showed that greater relative amyloid plaque clearance was correlated with greater clinical benefit. In June 2021, Lilly announced the U.S.
government for its neutralizing antibody therapies authorized for emergency use as a treatment for COVID-19. Bamlanivimab and etesevimab together and bamlanivimab alone have not been approved by the FDA for any use. who require oxygen therapy due to COVID-19, OR.
Food and Drug Administration (FDA) has cleared the Company’s Investigational New Drug (IND) application and that the United Kingdom Medicines and Healthcare Products Regulatory Agency (MHRA) has authorized its Clinical Trial Application (CTA) for a Phase 1/2 clinical trial evaluating INZ-701 in adults with ENPP1 deficiency.
As UNITY reported in its press release, this “Phase I, first-in-human, open-label, single-ascending dose study being conducted by UNITY is designed to evaluate the safety, tolerability, and pharmacokinetics of UBX1325 in patients with advanced DME. SUZHOU, China and ROCKVILLE, Md. , 22, 2020 /PRNewswire/ — Ascentage Pharma (6855.HK),
Food and Drug Administration (FDA) revoke the Emergency Use Authorization (EUA) for bamlanivimab (LY-CoV555) 700 mg alone. for the treatment of COVID-19 – as planned with the FDA – follows the modification of contracts with the U.S. Bamlanivimab and etesevimab together have not been approved by the FDA for any use.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.











































Let's personalize your content