This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
Still, more than 90 percent of drug candidates fail in clinical trials, with even more that never make it to the clinical stage. Together, the tools estimate how a drug may impact diverse outcomes of interest to drug developers: general cellular health, pharmacokinetics, and heart and liver function.
Regulatory bodies such as the FDA oversee clinical trials to ensure that studies’ design, conduction, analysis, and reporting are per established guidelines and laws. Any delays or missteps in bioanalysis during a Phase I trial can derail the trajectory of a promising drug.
Authors: Matt Cooper, PhD, Executive Director, Therapeutic Strategy Lead, Oncology; Megan Morrison, Vice President, Asia Pacific Strategy Lead Adaptive trial designs have become essential in oncology, offering a flexible and efficient approach for conducting clinical trials.
In November 2023, at Outsourcing Clinical Trials Dach in Zurich, our Executive Director, Oncology Strategy Lead, Matt Cooper , presented “Delivering Oncology Studies – Challenges and Considerations.” Umbrella Trial: Examines numerous drugs administered as individual drugs or combinations in a single tumor type.
Table 1: Small molecule drugs approved by the FDA in 2023 with reported involvement of phase II mechanisms In vitro : In vivo differences Incubation of the SGLT2 (sodium-glucose co-transporter-2) inhibitor bexagliflozin in human liver microsomes points to metabolism through both oxidation and glucuronidation to 6 main metabolites.
The FDA has approved two vaccines, and three vaccines are available for emergency use, to prevent COVID-19 and the serious clinical outcomes associated with COVID-19, including hospitalization and death. The FDA urges the public to get vaccinated and receive a booster when eligible. The FDA granted approval to Gilead Sciences Inc.
AI also has the potential to incorporate real-world data (RWD) obtained from electronic health records (EHRs), medical claims or other data sources to inform the design and optimization strategy of clinical trials.
of new cancer drugs tested in Phase I were likely to receive Food and Drug Administration (FDA) approval. Innovation Organizations conducting oncology clinical trials face challenges distinct from the rest of the research community. Typical clinical development timelines for anticancer drugs average an estimated 6.7
Food and Drug Administration (FDA) ’s focus appears to be on Emergency Use Authorizations (EUAs) for the Pfizer-BioNTech and Moderna COVID-19 vaccines, as the year wraps up there are still some PDUFA dates on the agency’s calendar. We look forward to working with the FDA to bring ABP 798 to market.”. Although much of the U.S.
The study demonstrated favorable proof-of-concept for LYT-100’s tolerability and pharmacokinetic (PK) profile, which will also enable twice-a-day (BID) dosing of LYT-100 in future studies. The therapeutic dose of pirfenidone approved by the US Food and Drug Administration (FDA) for the treatment of IPF is 801 mg three times a day.
However, when it comes to an IND and supporting a clinical trial, FDA’s primary focus is on healthy volunteer and patient safety. It is critical that the nonclinical program outlined in the PIND briefing document is presented in a manner that allows FDA to provide relevant input on the required IND-enabling studies.
Food and Drug Administration (FDA) in terms of PDUFA dates. Here’s a look at some of the upcoming target action dates on the FDA’s calendar. . The company is also still conducting a Phase III trial in the adult ADHD patient population. The sNDA was built on data from the Phase III CANDOR trial.
A weekly update on new drug approvals and indications from the US Food and Drug Administration (FDA). . It also provided supporting pharmacokinetic data demonstrating the opioid antagonist’s safety and efficacy. . controlled trial had not received previous systemic therapy for metastatic disease. New approvals. blind, placebo?controlled
While the type, number, and design of these studies vary based on product-specific characteristics, IND-enabling packages submitted to the FDA generally include key information about the pharmacology, pharmacokinetics, and toxicology of the product. All these studies need to be performed under GLP.
The US FDA Modernisation Act 2.0., puts an end to the previous mandate that all drugs need to be tested on animals prior to human clinical trials. These alternative models for drug testing are promising but are relatively at their infancy with need for rigorous optimisation and standardisation of protocols to reach FDA standards.
The first patients have been enrolled in a phase 1 randomized placebo-controlled clinical trial to study a therapeutic vaccine for opioid use disorder developed by researchers at the University of Minnesota Medical School.
FDA updates guidance on developing drugs for Covid-19, replacing pandemic-era version Last week, FDA published the third update to its guidance on the development of products to prevent or treat Covid-19. The FDA also implemented new flexibilities for certain regulated products and processes, typically via enforcement discretion.
Since 1962, the FD&C Act has authorized FDA to require that sponsors of clinical trials submit data from “preclinical tests (including tests on animals)” in order to demonstrate that their drug is safe enough to advance to testing in humans. For more on FDORA’s other provisions, see HPM’s complete summary here ). 21 U.S.C. §
United Kingdom Medicines and Healthcare Products Regulatory Agency authorized Clinical Trial Application.
With these important regulatory clearances for our first-in-human clinical trial for INZ-701 in subjects with ENPP1 deficiency, we have transitioned from a research-stage to a clinical-stage company.
BOSTON, Jan.
A pivotal Phase 3 trial evaluating Dupixent ® (dupilumab) for the treatment of children aged 6 months to 5 years with moderate-to-severe atopic dermatitis, a chronic type 2 inflammatory disease, met its primary and all secondary endpoints. Yancopoulos, M.D., President and Chief Scientific Officer at Regeneron. “ In 2016, the U.S.
“Population pharmacokinetic/pharmacodynamic modelling of eplontersen, an antisense oligonucleotide in development for transthyretin amyloidosis” British Journal of Clinical Pharmacology. S2CID 250989659. ^ “Eplontersen: FDA-Approved Drugs” U.S. Food and Drug Administration (FDA). Ionis Pharmaceuticals.
Altasciences Completes Successful Phase I Trial of Ischemix’ Novel Compound for Treatment of Traumatic Brain Injury (TBI) pmjackson Mon, 08/28/2023 - 14:04 Laval, Québec, August 30, 2023 – Altasciences is pleased to have completed a Phase I trial on Ischemix, Inc.’s We are proud to support Ischemix in advancing this vital new therapy.”
The FDA EUA submission is based on an interim analysis of efficacy and safety data from the Phase 3 COMET-ICE (COVID-19 Monoclonal antibody Efficacy Trial – Intent to Care Early) trial, which evaluated VIR-7831 as monotherapy for the early treatment of COVID-19 in adults at high risk of hospitalisation. About COMET-ICE.
Phase III BRIDGE open-label, switch-over clinical trial met key objectives for safety and efficacy.
Food and Drug Administration (FDA) for pegunigalsidase alfa for the proposed treatment of adult patients with Fabry disease via the FDA’s Accelerated Approval pathway.
CARMIEL, Israel and BOSTON , Dec.
18, 2021 /PRNewswire/ — Genkyotex SA , a subsidiary of Calliditas Therapeutics AB (publ) (“Calliditas”) (Nasdaq OMX – CALTX; NASDAQ – CALT), today announced positive Phase 1 data demonstrating a favorable safety and pharmacokinetic profile of high-dose setanaxib, Genkyotex’s lead asset. STOCKHOLM , Jan.
FDA looking to beef up its remote assessments with artificial intelligence According to a new FDA contract notice, the agency is interested in procuring a new artificial intelligence-based Optical Character Recognition system to extract data from PDFs obtained during remote regulatory assessments. What does the FDA want to do, anyways?
In the absence of a clinical trial result or FDA label to point to, how does one create the case and target product profile (TPP) around a new target? Trial considerations will ultimately inform nonclinical studies such as GLP tox, and will serve as the basis for timeline and budget discussions around a fundraise.
Phase I Trial. phase I trial is a randomized, double-blind, placebo-controlled, single and multiple dose escalation study to evaluate safety, tolerability, pharmacokinetics and pharmacodynamics (biomarkers – FGF19 and C4) of ASC42 in healthy subjects. This trial also investigates the food effect on ASC42 exposure.
The trial, which will explore the pharmacokinetics and safety of ATX01 in healthy volunteers, is due to start in January 2021. In parallel, AlgoTx firmed-up ATX01’s development pathway via a pre-IND consultation with the FDA and obtained an Orphan Drug Designation from the FDA to explore ATX01’s activity in erythromelalgia.
FDA offers a status check on its diversity in research provisions, one year post-FDORA At a workshop on FDA’s implementation of new statutory requirements for diversity in clinical research, agency and industry representatives gave a status update on implementation.
The NDA for mobocertinib is primarily based on results from the Phase 1/2 trial , which is evaluating the safety and efficacy of oral mobocertinib in patients with mNSCLC. The application was submitted under the FDA’s accelerated approval program. About the Phase 1/2 Trial. In 2019, the U.S. About EGFR Exon20 Insertion+ mNSCLC.
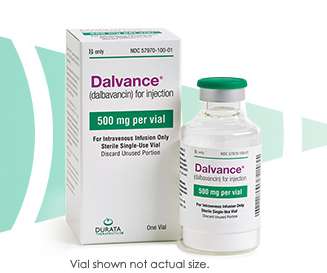
Food and Drug Administration (FDA) approved DALVANCE® (dalbavancin) for the treatment of acute bacterial skin and skin structure infections (ABSSSI) in pediatric patients from birth. The trial was not powered for a comparative inferential efficacy analysis. AbbVie (NYSE: ABBV) today announced that the U.S.
There are over 2,000 CGTs currently being evaluated in clinical trials, with more than 200 in Phase III and 10-20 per year estimated to come to market by 2025. Food and Drug Administration (FDA). Working with experienced providers can help clients build data sets that satisfy current and future FDA requirements.
Food and Drug Administration (“FDA”) for the treatment of agitation associated with delirium. The Company plans to initiate a Phase 2 trial within the next several months. “We The Company also plans to initiate a Phase 2 trial in hospitalized patients suffering from agitation associated with delirium within the next several months.
On July 31, 2024, the US Food and Drug Administration (FDA) announced Fiscal Year 2025 (FY2025) Prescription Drug User Fee Amendments of 2022 (PDUFA VII) fee rates for the review of human drug and biological product applications along with prescription drug program fees. FDA-2024-N-0007. FDA-2023-N-2850. 89 FR 61474.
A review of FDA’s Postmarketing Requirements and Commitments database reveals that one of the most common reasons FDA requires postmarketing studies is to assess the impact of a drug on maternal and fetal outcomes when taken by pregnant women. Additionally, this review excluded pharmacokinetic and animal studies.
FDA’s newest draft guidance lays out considerations for developing treatments for diabetic foot infections In a new draft guidance document released within weeks of a related clinical practice guideline, the FDA provides considerations for developing therapies to treat diabetic foot infections, focusing on Phase 3 efficacy trials.
The Phase 2/3 trial is an open-label, multi-center, single-arm study in approximately 140 pediatric participants under 18 years of age. We are proud to expand studies of our novel COVID-19 treatment to include pediatric participants to further evaluate the safety and efficacy of this treatment in this important population.”.
Food and Drug Administration (FDA) has approved long-acting atypical antipsychotic INVEGA HAFYERA™ (6-month paliperidone palmitate), the first-and-only twice-yearly injectable for the treatment of schizophrenia in adults. DFAPA, Medical Director at ATP Clinical Research and 6-month paliperidone palmitate clinical trial investigator.
BY AMANDA CONTI, ALEXANDER GAFFNEY, MS, RAC SEP 18, 2023 9:24 PM CDT Background: FDA’s standards for evidence When seeking approval from the FDA, companies are required to demonstrate that their product is safe and effective when used as intended. In one landmark case, Warner-Lambert Co.
The FDA and oncology: 2023 year in review As we round the corner into the last few weeks of 2023, AgencyIQ has taken a look back at a very busy year for the FDA’s oncology staff, and for sponsors. Since its inception, OCE has initiated an extensive list of initiatives and has taken the lead on over forty guidances published by FDA.
The open-label Phase 2a ‘AMBITION’ study is designed to assess safety, tolerability, pharmacokinetics and biomarker analyses for early assessments of efficacy of 75 mg and 225 mg CRV431, administered orally to F2 and F3 NASH patients (n=18/dosing group), once daily for 28 days.
Food and Drug Administration (FDA) has extended the Prescription Drug User Fee Act (PDUFA) date for review of the Company’s Biologics License Application (BLA) seeking accelerated approval of pegunigalsidase alfa (PRX–102) for the proposed treatment of adult patients with Fabry disease.
About Pegunigalsidase Alfa.
The AMBITION trial is the first placebo-controlled study of CRV431 in NASH patients with evidence of moderate-to-severe fibrosis. The primary objectives of the AMBITION trial are to assess safety and tolerability of CRV431, as well as to delineate pharmacokinetics. To date, there are no approved drugs to treat NASH.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.











































Let's personalize your content