This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
A group of independent experts wasn't convinced by clinical trial data from company Lykos Therapeutics, which is seeking FDA approval of MDMA-assisted treatment for post-traumatic stress disorder.
Expert advisers to the agency voted 17-1 that Brainstorm's clinical trial data did not show the company's stem cell treatment was effective for treating ALS.
Expert advisers to the agency voted 17-1 that Brainstorm's clinical trial data did not show the company's stem cell treatment was effective for treating ALS.
The decision makes Elevidys available to Duchenne patients at least 4 years of age, despite mixed trial results that have led to skepticism about its effectiveness.
Food and Drug Administration (FDA) plays a pivotal role in fostering the development of treatments for rare diseases through its Orphan Products Grants Program. Each year, FDA selects a limited number of clinical trials to fund to help sponsors pursue development of medical products for rare diseases and advance their field.
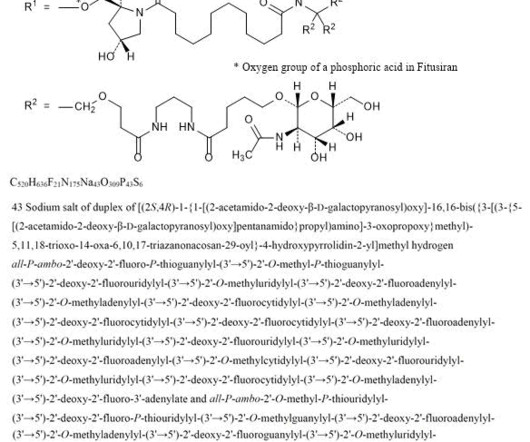
2] History The efficacy and safety of fitusiran were assessed in two multicenter, randomized clinical trials which enrolled a total of 177 adult and pediatric male participants with either hemophilia A or hemophilia B. [2] The FDA granted the approval of Qfitlia to Sanofi. Fitusiran 1711.0g/mol, Fitusiran 1711.0g/mol, 26 March 2025.
Last week DNA Science covered a setback in a clinical trial of a gene therapy for Duchenne muscular dystrophy (DMD). Also recently, FDA’s Cellular, Tissue, and Gene Therapies Advisory Committe turned down a stem cell treatment for amyotrophic lateral sclerosis, aka ALS, Lou Gehrig’s disease, or motor neuron disease.
As our understanding of the underlying biology of disease grows more sophisticated, emerging therapies operate on increasingly complex biopathological systems and mechanisms. A surrogate endpoint is a marker used in clinical trials as a substitute for a direct clinical outcome. There are several types of biomarkers to consider.
Elsewhere, Novo started up an online pharmacy and the FDA cleared testing of a base editing therapy for Duchenne. Phase 3 studies for Amgen’s closely watched obesity drug were posted on a federal database.
Valentine — On November 22, 2022, FDA approved CSL Behring’s BLA for Hemgenix (etranacogene dezaparvovec), an AAV-based gene therapy for the treatment of adults with Hemophilia B who currently use Factor IX prophylaxis therapy, have current or historical life-threatening hemorrhage, or have repeated, serious spontaneous bleeding episodes.
In an era where clinical trials are increasingly global, it’s more imperative than ever to leverage international expertise. Data and safety monitoring boards (DSMBs), also known as data monitoring committees (DMCs), play a critical role in overseeing a clinical trial’s safety and efficacy. local standards of care).
Importantly, the Hub is intended to establish a new model within FDA, which leverages cross-Agency expertise in providing guidance and conducting reviews for products for rare disease populations. By Sarah Wicks & James E. Valentine & Frank J. those reviewed by the CDER Division of Rare Diseases and Medical Genetics).
2] [6] Suzetrigine is the first medication to be approved by the US Food and Drug Administration (FDA) in this new class of pain management medicines. [2] The FDA has long supported development of non-opioid pain treatment. acting director of the FDA’s Center for Drug Evaluation and Research.
The number of pediatric patients diagnosed with nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH) is on the rise, yet there are currently no approved therapies to treat NAFLD and NASH in adult or pediatric populations. As therapies for the treatment of NASH in adult patients go this year to the U.S.
Food and Drug Administration (FDA)-approved therapies for treating PAH were primarily vasodilators, designed to overcome the imbalance between vasoactive and vasodilator mediators and to restore endothelial cell function. Their early definition — as well as plans for recording and tracking — is a major factor in a trial’s success.
FDA classifies it as a “nonsteroidal treatment” – not a gene therapy, but it affects gene expression. The New Drug Duvyzat (givinostat), a type of drug called an HDAC inhibitor, has been in clinical trials to treat cancers and other disorders of the blood, Crohn’s disease, and a form of juvenile arthritis.
Sasinowski — On March 21st, FDA announced the approval of the first nonsteroidal therapy for the treatment of Duchenne Muscular Dystrophy (DMD) (FDA press release available here ). The primary basis for approval, like other drugs for DMD, was based on a single placebo-controlled randomized trial. By Charles G.
The Food and Drug Administration (FDA) recently released new guidance regarding cellular and gene therapy products, one of which may significantly impact early-phase clinical trials of such products. The Benefits of the Umbrella Trial There are many potential benefits to the umbrella trial.
These therapies have broadened treatment options for patients to expand beyond the more traditional small molecule drug alternatives. Patients and caregivers also assess the benefits offered by different therapies, weighing the progression-free survival with their off-target effects. 3D rendering of Antibody Drug Conjugate Molecules.
NC: I think the continued growth in cell and gene therapy research and the approvals we’re seeing for genetically-driven rare diseases. First, the beginning of the FDA’s START program, with the goal of accelerating the development of novel drugs and biological products for rare diseases.
Read the Article Rare Roundup Bespoke Gene Therapy Consortium Publishes First ‘Playbook’ to Bring Rare Disease Programs to Clinic Gene-based therapy is still a new arena; even as scientists across the world work on gene therapies, a major question is how to best prepare these treatments for clinical trials and regulatory approval.
Food and Drug Administration (FDA) announced its acceptance of the Biologics License Application (BLA) for exa-cel. In recognition of the unmet need and medical urgency for innovative therapies in the sickle cell space, the FDA granted exa-cel Priority Review, with a formal decision expected by December 8, 2023.
s Mark Tobolowsky co-authored the peer-reviewed article “ Microdystrophin Expression as a Surrogate Endpoint for Duchenne Muscular Dystrophy Clinical Trials ” in the recently published edition of Human Gene Therapy. Hyman, Phelps & McNamara, P.C.’s
Designation is supported by Phase II efficacy and safety data that will be presented at ATS 2022 Boehringer Ingelheim plans to study this novel investigational therapy in patients with progressive fibrosing interstitial lung diseases. The FDA Breakthrough Therapy designation is supported by data collected to date.
Zidovudine, didanosine, and stavudine are FDA-approved NRTIs, while nevirapine, efavirenz, and delavirdine are FDA-approved NNRTIs. Several agents are in clinical trials, including apricitabine, racivir, elvucitabine, doravirine, dapivirine, and elsulfavirine.
Sponsors using master protocols to test new drugs and therapies for treating or preventing COVID-19 should base their analyses on comparisons between control arm participants who were concurrently randomized, the FDA advised in a final guidance released yesterday. Read the guidance here: www.fdanews.com/05-17-21-COVID-19.pdf.
Food and Drug Administration (FDA) issued two guidance documents outlining the necessary evaluations during the clinical development of oligonucleotide therapeutics: Clinical Pharmacology Considerations for the Development of Oligonucleotide Therapeutics and Nonclinical Safety Assessment of Oligonucleotide-Based Therapeutics .
The field of cell and gene therapies (CGT) is constantly evolving, and there has been significant progress in this area of research. However, despite the promise of these therapies, the regulations governing them lag the science, which in turn hinders the clinical translation of these novel medicines.
The newest FDA-approved gene therapy treats the severe, skin-peeling condition dystrophic epidermolysis bullosa (DEB). The gene treatment has been a long time coming, but it differs from the handful of other approved gene therapies: it isn’t a one-and-done. DEB has been a candidate for a gene therapy since 2002.
Accelerated approval is an expedited regulatory pathway designed to hasten the availability of drugs (including biologics) that treat serious conditions, offer advantages over existing therapies, and address unmet medical needs. In essence, these trials are the final step that turns provisional approval into full approval.
Novartis today announced the US Food and Drug Administration (FDA) has granted accelerated approval for Kymriah ® (tisagenlecleucel) for the treatment of adult patients with relapsed or refractory (r/r) follicular lymphoma (FL) after two or more lines of systemic therapy.
Though clinical trials are often discussed in the context of medications and therapies, the devices that administer these medications must also be approved by the FDA before they can be released to market.
This approval will allow some patients the option of receiving once-monthly injections in lieu of a daily oral treatment regimen,” said Dr. John Farley, director of the Office of Infectious Diseases in the FDA’s Center for Drug Evaluation and Research.
Food and Drug Administration (FDA) for the treatment of adult patients with deleterious or suspected deleterious BRCA -mutated ( BRCA m) metastatic castration-resistant prostate cancer (mCRPC). Patients should be selected for therapy based on an FDA-approved companion diagnostic for LYNPARZA. In the U.S.,
Roche announced that gantenerumab, an anti-amyloid beta antibody developed for subcutaneous administration, has been granted Breakthrough Therapy Designation by the U.S. Food and Drug Administration (FDA) for the treatment of people living with Alzheimer’s disease (AD).
She is scheduled to be the 2nd patient in the world to receive ASO therapy for KAND. March 2023 – We met with n-Lorem and found out Susannah’s ASO (1st KAND patient to receive ASO therapy for KAND) would work for Sloane. February 2024 – n-Lorem submitted an IND (investigational new drug) to the FDA for Sloane.
Advancing reduction of drug use as an endpoint in addiction treatment trials astewart Thu, 03/06/2025 - 09:59 Nora's Blog March 18, 2025 Image Getty Images/ SolStock This blog was also published in the American Society of Addiction Medicine (ASAM) Weekly on March 18, 2025.&
This process can be daunting, but understanding how to manage feedback effectively is crucial for developing and ultimately gaining approval for new therapies, especially in oncology clinical trials. An illustrative example of harmonization between agencies exists via the European Medicines Agency (EMA) and U.S.
Mr. Barrow has over a decade of experience leading drug development programs aimed at identifying and testing novel treatments in a wide range of disease conditions under FDA and EMA.
.
NEW YORK , Jan.
MindMed Co-CEO J.R.
About MindMed.
Kite’s Global CAR T-Cell Therapy Manufacturing Network Increasing Capacity by 50% to Meet Patient Demand for New Cancer Therapies. — Scalable and Adaptable Facility Provides Flexibility for Current and Future Cell Therapy Innovation. The site will produce Kite’s FDA approved CAR T-cell therapy used to treat blood cancer.
This can be converted to a clinical trial simulator, which can model a clinical trial before the trial design is finalized and initiated. AI approaches are optimizing trial designs With a significant increase in the number of regulatory submissions to the U.S.
. – Preclinical Data Underscore Treatment Potential for PBFT02 in Frontotemporal Dementia with Granulin (GRN) Mutations, a Devastating, Progressive Disorder Impacting Adults with No Approved Disease-Modifying Therapy Options. FTD is a debilitating form of early onset dementia that currently has no approved disease-modifying therapies. “We
Due to the less extensive clinical trial requirements and the competitive nature of the biosimilar market, these biologics can be produced and marketed at significantly lower prices than their reference counterparts. Food and Drug Administration (FDA) have been instrumental in shaping global regulatory frameworks for biosimilars.
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.









































Let's personalize your content