This site uses cookies to improve your experience. To help us insure we adhere to various privacy regulations, please select your country/region of residence. If you do not select a country, we will assume you are from the United States. Select your Cookie Settings or view our Privacy Policy and Terms of Use.
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Used for the proper function of the website
Used for monitoring website traffic and interactions
Cookie Settings
Cookies and similar technologies are used on this website for proper function of the website, for tracking performance analytics and for marketing purposes. We and some of our third-party providers may use cookie data for various purposes. Please review the cookie settings below and choose your preference.
Strictly Necessary: Used for the proper function of the website
Performance/Analytics: Used for monitoring website traffic and interactions
By harnessing the vast amounts of data generated throughout the development pipeline, pharmaceutical companies can accelerate the discovery of novel therapies, optimize clinical trial design, enhance drug safety monitoring, and deliver personalized medicine, ultimately improving patient outcomes and transforming the future of healthcare.
The current version of Chemistry42 uses over 40 generative models, including generative autoencoders and generative adversarial networks as well as both structure-based and ligand-based drug design to generate and optimise de novo smallmolecules. The novel molecules were further ranked based on their ADME and selectivity profiles.
In a brief section entitled “Predicting Protein-SmallMolecule Complexes”, the authors mention their efforts to generate structures of bound non-covalent and covalent smallmolecule ligands. from the deposited model. Similar issues can impact datasets used for QSAR or ADME modeling.
Now >20% of all commercialised medicines in the pharmaceutical industry contain a fluorine atom [2]. Even newer from the pharma benches are 12 smallmolecule drugs highlighted by Chris de Savi [4], whose structures were first disclosed at the ACS and AACR meetings in Q1 2023. Benjamin M. Airaksinen.
Traditionally, drug discovery has focused on small-molecule therapeutics, typically with a molecular weight of less than 500 Daltons. 2 Typically, small-molecule drugs target active sites buried inside proteins. What are macrocycles and why are they interesting for drug discovery?
We organize all of the trending information in your field so you don't have to. Join 15,000+ users and stay up to date on the latest articles your peers are reading.
You know about us, now we want to get to know you!
Let's personalize your content
Let's get even more personalized
We recognize your account from another site in our network, please click 'Send Email' below to continue with verifying your account and setting a password.



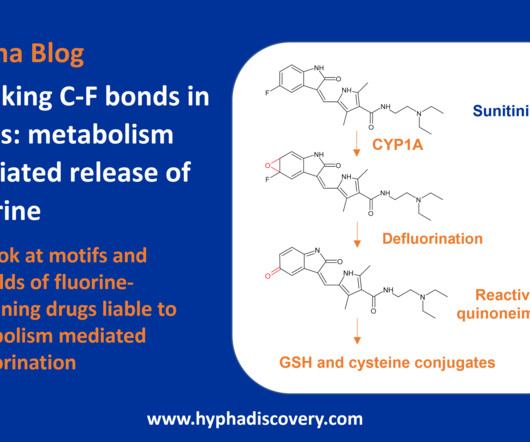







Let's personalize your content